

A deep dive on TypeIIs Restriction enzymes
The Workhorses of Genomics


This is a post to help inform people on the various tools used in synthetic biology and DNA sequencing. I hope to get you comfortable with downloading DNA sequences and evaluating them for Restriction enzyme sites so you can visualize the restriction maps being discussed in this recent preprint (Bruttel et al). It is an important preprint and is the most credible explanation of what might have occurred in a lab somewhere playing with these viruses. You will need a free piece of software known as SnapGene. I hope these tools will give you some familiarity with the picks and shovels of the industry and allow you to decide for yourself if you think this virus was engineered or randomly evolved in nature.
Endonuclease fingerprint indicates a synthetic origin of SARS-CoV-2
DownLoad SARs-CoV-2.fasta and load it into SnapGene
Hit the New button in the upper left and it will guide you through loading the SARs-CoV-2 genome into SnapGene.
Then click on the next button down from New that has a SalI over cut site. Its the first button on the top of the vertical side bar. It will give you a menu which allows you to look at many libraries of restriction enzyme from NEB. Select the Golden Gate option.
This will select 9 enzymes to look at but the most popular ones used are BsaI and BsmBI. Just check those two and you’ll get a restriction map of where these are in the SARs-CoV-2 genome.
You will need to select on the Enzymes tab shown in blue at the bottom of the screen.
Then click on the Map icon on the bottom bar.
This will show you the TypeIIs restriction map for BsmBI and BsaI in SARs-CoV-2. Does this match Bruttel et al?
Looks legit.
Now you have the tools to load in any other Coronavirus genome and double check their restriction maps. Here is RatG13.
So what is all this hubub about restriction sites? They are short DNA sequences that occur quite frequently in genomes. These short sequences are like homing beacons that certain enzymes home in on like a low signature zip code. You can ‘guestimate’ the frequency of occurrence of any DNA sequence with some shorthand math keyed to its length. 4^N is the number of sequences a sequence can code for with said length N. A DNA sequence of 4 letters can have 256 different potential sequences (4^4). 5 bases are 1024 (4^5) and 6 bases long are 4^6 or 4096 sequences. So a quick way of estimating how many 6 base recognition sequences might exist in 30kb is to divide the target genome by the frequency of the cutter (6 cutter = 4^6). You should expect to see about 7 sites by chance as a sequence of 6 bases in length should occur once every 4096 bases and 30Kb/4096 = 7.32. While there should be 7 sites by chance in SARs-CoV-2, their distribution should be semi-random. This is one of the key aspects of the papers argument: How departed from random are these sites and what do other CVs look like in this regard.
What do restriction enzymes do once they home in on their restriction sites? These are enzymes that recognize a particular sequence and cut the DNA at that site.
Most cats behave like TypeIIs restriction enzymes. They sit on your keyboard and cut things far away from the letters they sit on.

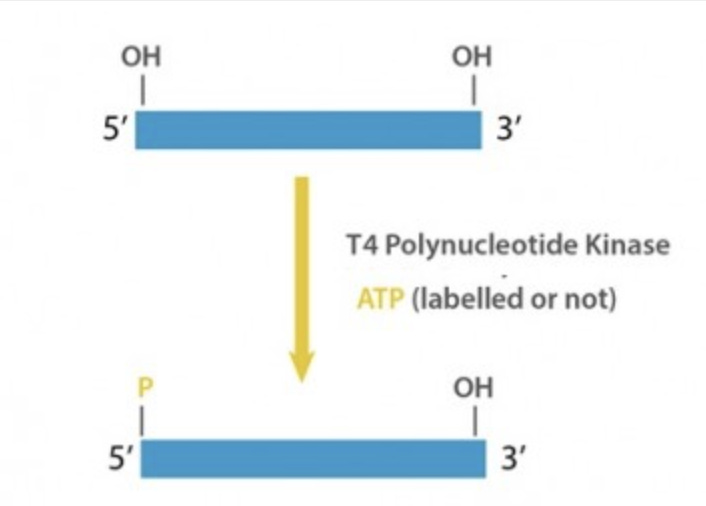
The arrows in the below NEB restriction enzyme description depict where the DNA will be cut. EcoRI has a 6 base recognition sequence of GAATTC and when it cuts it will leave a 5’ overhang of 5’PO4..AATT-3’. The 5’PO4 (phosphate) is not shown in the NEB depiction but it is an important feature as without it you can’t glue the DNA to anything. This gluing is called ligation and it needs a 5’PO4 to link to a 3’OH group. Like male and female connectors on a cable, DNA is directional and ligases need both groups to seal the strand covalently.

Some fragments of DNA are missing 5’PO4 groups. This often happens when someone PCRs a fragment of DNA. Unless the PCR primer used has a 5’PO4 group on it (rarely done), the PCR products will be formed with dephosphorylated 5’ ends but the fragments will have active 3’OH groups left by the polymerase extension of nucleotides.
NEB has a good video describing 5’ overhangs and 3’ overhangs and the various enzymes use to blunt them and phosphorylate them. T4 Kinase is often used to put Phosphates on PCR products in a process also known as End-Repair.
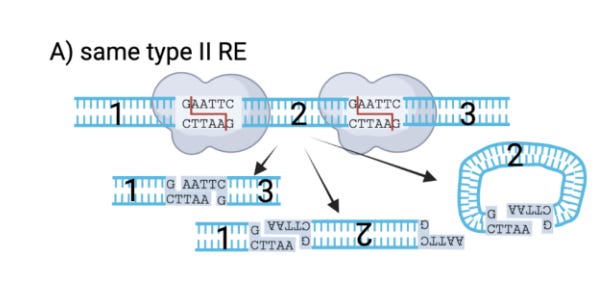
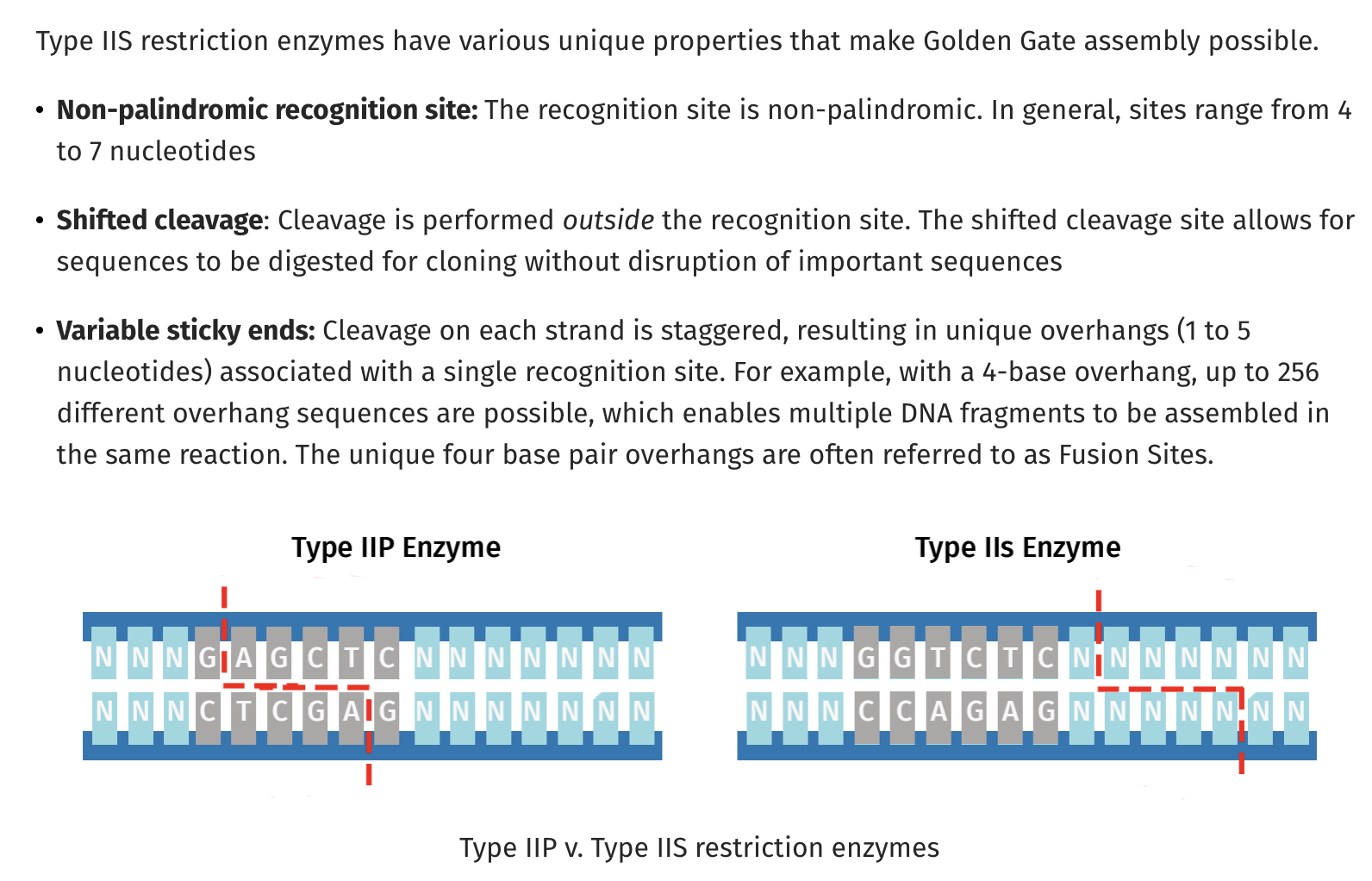
One limitation of old fashion restriction enzymes like EcoRI is that they cut within their recognition sequence and are often palindromes. This GAATTC is palindromic sequence as its reverse complement is also GAATTC. EcoRI reads the same on the top strand 5’→3’ as it does on the bottom strand. For those into musical palindromes I recommend UFO TOFU from Bela Fleck.
Note, since the DNA overhang that is left behind (AATT) is palindromic and self complementary, this means the addition of DNA glue (ligase) will make many products as the ends of all fragments match each other.
This is depicted in Bruttel et al. in Figure 1A. The molecules can ligate to each other or with inversions or even into circles/mobius strips. So if you want cut things and randomly glue them to each other in every combination possible, this approach is perfect but if you want to direct the organized ligation of fragments in a particular series you need to get a bit smarter.
TypeIIs restriction sites become very handy here. They also have recognition sequences like TypeIIP restriction sites (EcoRI) but they cut distal to their recognition sequences. A very popular one used for making mate-paired next generation sequencing libraries utilized MmeI. If you had a short read sequencer and you only wanted to sequence the very ends of millions of very large pieces of DNA (1-40kb), you want a way to snip off the ends of this DNA and just sequence those while retaining the information that these ends were linked 20kb apart in their original form. These ends are called mate-pairs. This was required at the time as there was no way to universally PCR amplify 20kb pieces across the human genome in 2006 and long read next generation sequencing had yet to deliver on the success they have today.

This was core to one of the ways to SOLiD sequence large molecules of DNA. One would randomly shear your DNA to be sequenced (from 100Mb chromosomes into 20kb). You then size select a 20kb portion of the DNA with a gel. The shearing process leaves wide size distributions and you really want a tight distribution of sizes to be able to leverage the information of the mate-pairs being an expected 20kb apart. One would then End Repair this DNA after shearing because random shearing leaves a mixture of blunt ends and overhang ends of DNA.
Once blunted, the DNA can be ligated into a circle or be ‘circularized’. If you add in just the right ratio of insert DNA you can ligate this 20kb DNA into a circle with a microplasmid backbone (100bp) or a synthetic ‘internal adaptor’. If this backbone has a two terminally placed TypeIIs restriction sites like MmeI, you get a circular molecule with a ~100bp insert that has TypeIIs Cut sites placed at the edges of the 20kb fragment. If you cut with these enzymes the terminal 20bp of the 20kb fragment will be liberated and glued to the synthetic insert. This synthesized 100bp linker sequence also has a Biotin on it. Biotins are like molecular fish hooks you can use to pull out all pieces of DNA with streptavidin beads. Steptavidin is a protein that strongly binds biotin and you can purchase streptavidin coated magnetic particles that can isolate any biotinylated piece of DNA that have the linker ligated to it. So the linker sequence has your 20bp ends of the 20kb fragment covalently ligated into a fragment of DNA that has a fish hook on it. This becomes easy to then ligate sequencing primers on the termini of these excised ends and PCR amplify a sequencing library that contains millions to billions of ends of 20kb fragments of DNA. This is very powerful tool for genomics.
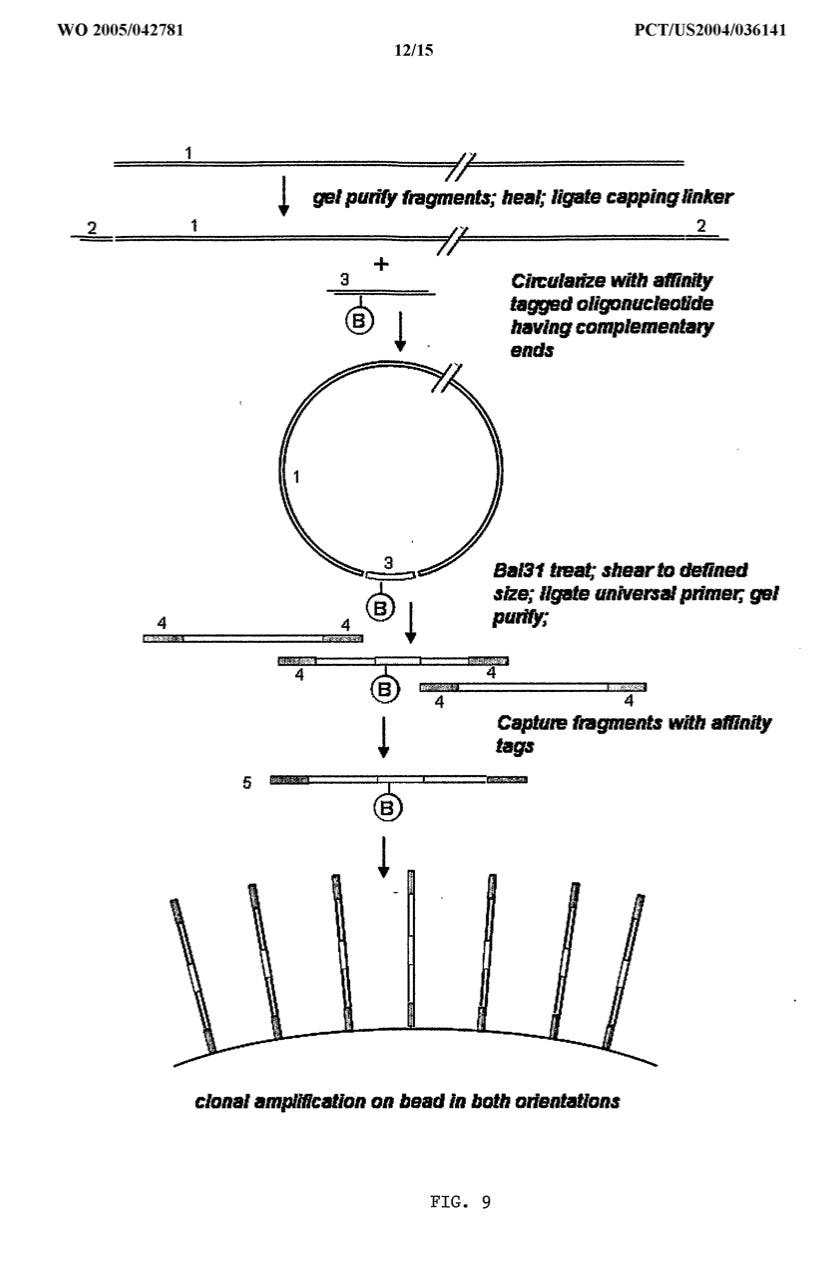
At the end of this procedure, you end up with just the terminal 20bp of 20kb fragments that are now short enough (under 500bp) to fit onto a next gen sequencer. Sequencing the ends of billions of molecules where the sequences are a known 20kb apart is very helpful for putting together genomes that have repeat structures that are longer than your sequence read length. They are very powerful tools for identifying large structural variations in genomes.
This technique was used by Johns Hopkins University to hunt for Personalized biomarkers of circulating Tumor DNA in patients blood. This made the cover of Science Translational Medicine.

Eventually this technique was improved with the use of a TypeIII enzyme that cut even larger pieces of DNA off the ends of 20kb DNA or what is often called High Molecular Weight (HMW DNA). The Enzyme EcoP15i cuts 25-27bases away from its restriction site and this produced sequencing tags that had better placement (more signature) in the human genome.

These were also called Jumping libraries and as the next gen sequencing read length improved even EcoP15i digests were too short at 27bp. To further improve this biotinylated Synthetic inserts were made that had recognition sites for Nicking enzymes. Nicking enzymes are just half ass restriction enzymes. They only cut one strand of the DNA. This can be very handy if you want a polymerase to take off from the 3’OH at the cut site and fill in the nick. When polymerases do this they peel away the strand that is in their way for extension much like a cattle prod on a train. You need a polymerase that has Nick translation activity (5→3 exonuclease activity) but these are easy to find. It’s really a shame they called this Nick translation as there is no translation to proteins involved.
Nick transcription would have been a better name for this procedure but as you will come to learn about mol bio… every thing has 3 names to keep outsiders feeling dumb and confused. Primers, Oligos and Probes… Plasmid, clones and Vectors. These are names for the same thing but what you choose to do with them changes their name. Primers are Oligonucleotides but suggest your intention to meet them with a polymerase. Oligos are just primers you ask someone else to make. They magically become primers when you use a polymerase and magically become a probe if you intend to shine a light on them. Vectors are plasmids you plan to put into something and clones are plasmids you plan to loose in a freezer somewhere.
This polymerase simply copies the strand that the half ass Nicking enzyme cut and effectively moves the Nick at ~20 bases a minute. A well timed Nick Translation reaction will push the nick 100-200 bases on average in a few minutes across a diverse library of molecules. You can speed this Nick translation rate up if you don’t starve it of nucleotides.
Now all you need is an enzyme that cuts the opposite side of a Nick and S1 nuclease is ideal for that job. Now we have a few hundred bases on either end of the 20kb molecule and we can sequence longer reads (longer than 25/27bp from EcoP15i) off each end.
These are handy tools to break apart genomes and sequence them with long range information. This is preferred over sequencing short fragments of DNA as you have more information to navigate complicated repeat structures that paired end sequencing from just 300bp inserts never can resolve. So Mate-Pair sequencing implies long jumping libraries and paired-end sequencing is usually derived from short fragment libraries under 1kb in size.
So how does one use these tools to synthesize new genomes from scratch?
This is actually much harder. We have great tools for reading DNA but we have very poor tools for writing DNA. We can Synthesize 100-150 base pair stretches of DNA with oligo synthesizers. These small ‘oligos’ must then be stitched together with ligases and since our word size is very short, we have many pieces that need to get glued together and we cant afford for 2/3rds of the molecules to ligate into circles or in the wrong orientation with each other. We need a mechanism to direct the stitching in a sequence directed manner.
Leave it to the Nobel Laureate Ham Smith. He won the Nobel prize for discovering restriction enzymes so he knew a thing or two about how to use them to assemble the first living organism genome in 2008. This isn’t just a 30kb walk in the park genome. This is a 582Kb microbial genome! In 2008!
So when people tell you its hard to make these SARs genomes, just know they were doing genomes 20 times bigger over a decade before the pandemic.
Note some of care taken in constructing this.

They also stitch oligos into 5-7kb fragments they can clone into bacteria for propagation or use PCR to amplify and sequence QC each piece. They can correct errors in bite size 5-7kb chunks and then assemble into 1/8th and 1/4 genomes. The SARs team could stop at the 30kb assembly and not perform the larger 582kb assembly as the SARs genome is only 30Kb. The bigger genomes require this hierarchical assembly approach. Much of this staged approached is predicated on the limitations of PCR. 20Kb PCR is hard to achieve. 5-7Kb is easy, robust and delivers plenty of product so the fragments of the genome are left in a size range that is easy to PCR modify without having rebuild everything from the base. It is like Modular indexing.
But notice they knocked out the pathogenicity, placed in a selectable marker and also added additional sequence to watermark the organism with the authors signature (coded in DNA). So if this puppy ever escapes it has dog tags and is attenuated.

This is the way.
There are several different ways to employ TypeIIs cutter in the assembly of these large fragments. One popularized technique is known as Golden Gate assembly.
So to Refresh TypeIIP enzymes cut internal to the recognition site and TypeIIs cut distal to their recognition sequence and if you place the TypeIIs sites at the ends of PCR primers, you can design your PCR products to be digestible where the TypeII site is the leaving group. It cuts itself off and leaves you with DNA fragments with sequence overhangs you can program. These are the NNNNN Sequences below. You can make those anything you want, and if you’re smart and lazy, you’ll pick sequences which direct the ligation to occur with only one outcome.
One end of Fragment 1 will have overhangs that only match one end of Fragment 2 and the ligases will not glue DNA overhangs together that do not match. The ligases are so discerning about the fidelity of these overhangs that we built an entire next generation sequencer that relied on the fidelity of this reaction. SOLiD sequencing was one of the most accurate DNA sequencers ever constructed. It suffered from shorter reads (50-75bp) but it had unmatched accuracy due to the nature of ligases and some very clever error correction codes built into the dye labeling scheme of the ligation oligos.
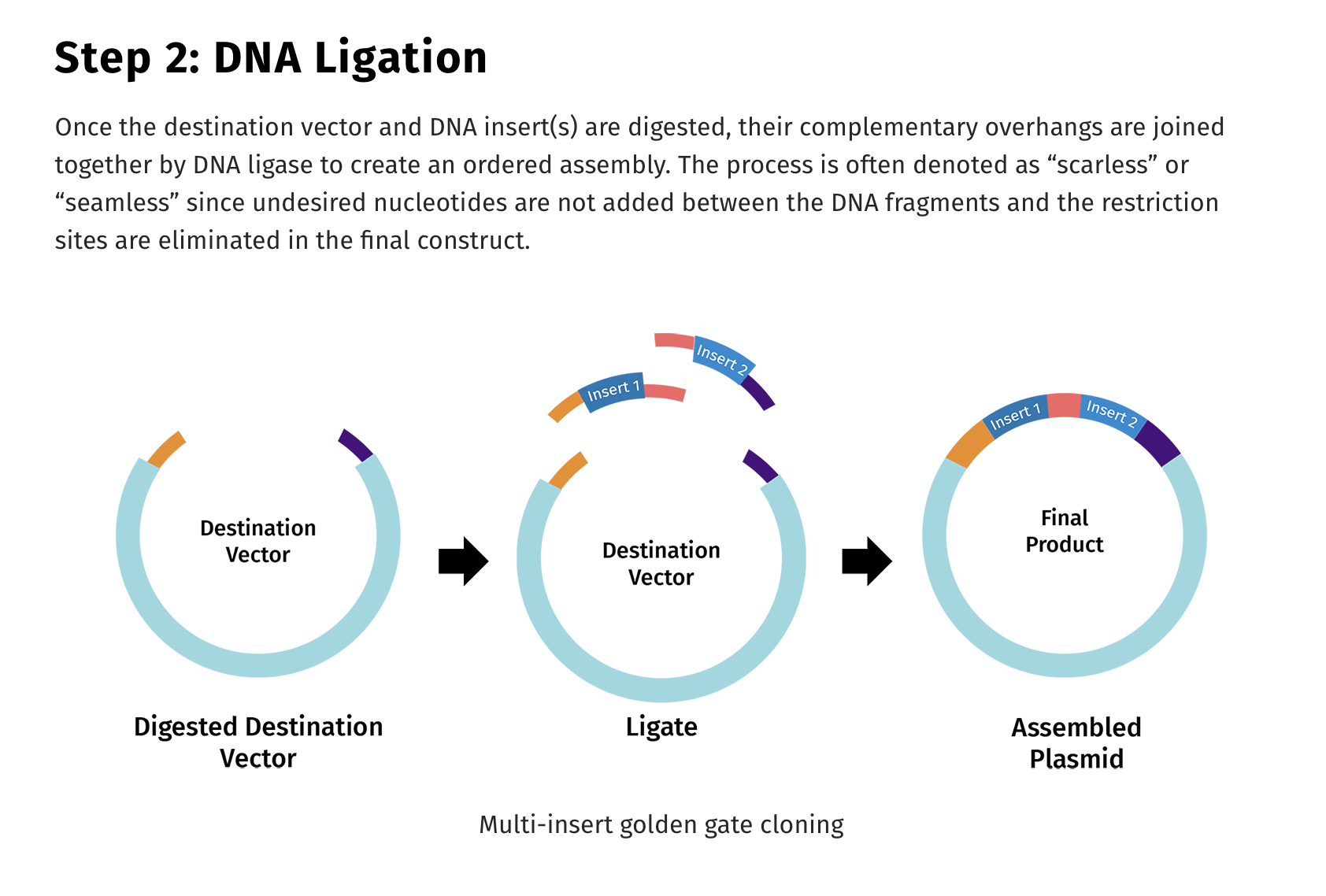
Here is where all the controversy around Bruttel et al seems to lie. In most depictions of Golden Gate, the restriction enzymes are the leaving group. They don’t end up in your assembled genome. But this is entirely up to the engineer. You simply flip their orientation and you can use them to liberate your 5-7kb fragments from a cloning vector or you can have them stay behind in your cloning vector. You choose. In this manifestation, the leaving group is you viral insert.
This is well described in Potapov et al.
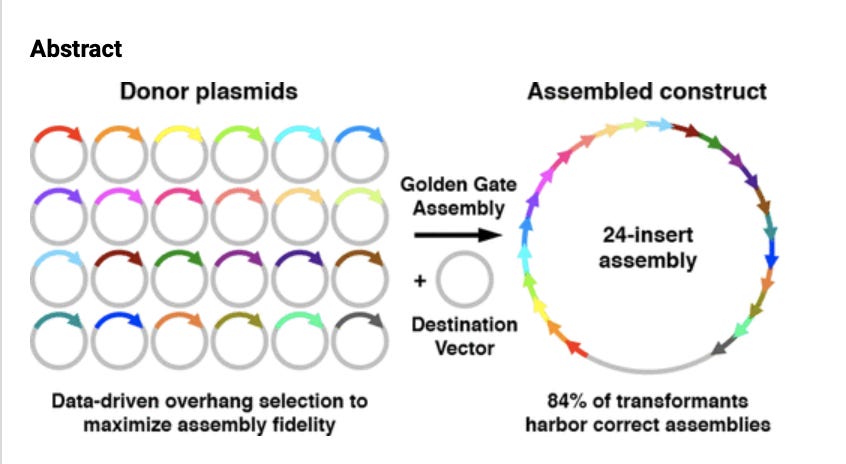
So why would the group that build SARs-CoV-2 want to leave TypeIIs cut sites behind in the genome when Golden Gate also enables you to make them the leaving groups (also known as No-See-Ums)?
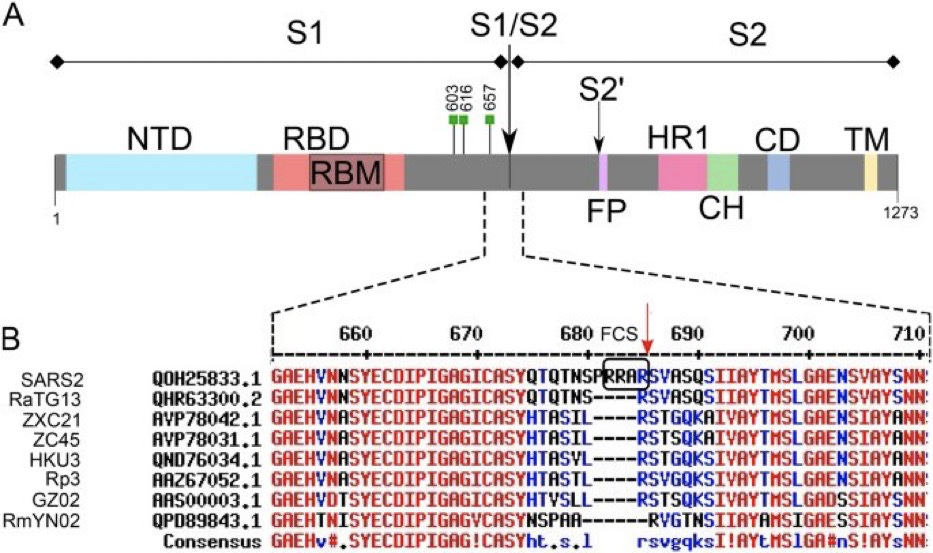
Well, what if you want to go back and change something? Like insert an FCS site to see if it changes host range? How do you do this if you don’t have some pitons left behind to anchor into?
You would need to excise the insert that contains the FCS from the storage plasmid. You’ll note the TypeIIs restriction sites that flank the FCS create fragment E which is pretty big. So if you want to modify just a tiny portion of this what do you do?
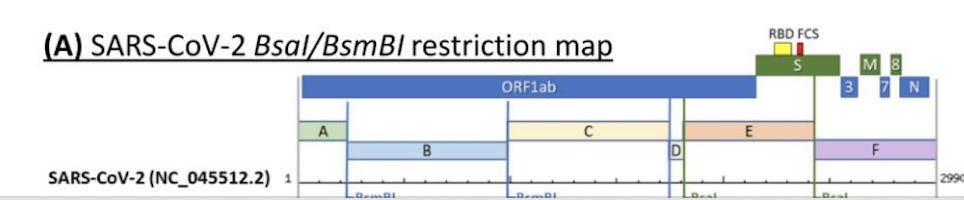
Fortunately the FCS has its own unique TypeIIs restriction site known as BsaXI.
BsaXI will chop Fragment E into 4 large pieces and 3 very small pieces. Do any of these sites make it easier to index to the FCS?

Well look at that! BsaXI perfectly excises the FCS and the SEB domain (YQTQTNSPRRA). Thats pretty damn lucky. But after cutting Fragment E into 4 large pieces and 3 small BsaXI pieces, how will you put humpty dumpty back together again?
Once again, the TypeIIs nature of these enzymes allows us to direct the reconstruction of these smaller fragments by coding the Ns in the overhang cut sites to only re-assemble in a desired order. What is also pretty handy about BsaXI is that it has lots of Ns on both sides (See NEB depiction above) of its recognition sequence (highlighted in red above). This means you could ask an oligo shop to synthesize anything you like there. If you want to test out a super-antigen with structural homology to the SEB bioweapon, you can easily do this. In fact, they did it with YQTQTNS sequence within the Ns of the BsaXI recognition site.
You could also use this design as a screening tool which asks the oligo synthesis facility to makes the new BsaXI insert to keep the Ns as Ns during synthesis (all combinations of ATCG). This would generate a combinatorial library of different BsaXI inserts one could insert and test all variants of SEB or the FCS and screen for which inserts are most functional or super-antigenic.
How would you prove these BsaXI sites are designed for overlap re-assembly? One just needs to look and see if the overlaps are incompatible and can only assembly in one direction.
So what do the other BsaXI sites leave as overhangs?
SEB/FCS BsaXI site has ATA and GCA overhangs.

Neighboring BsaXI sites have CGA and GAT (not complimentary to other BsaXI sites in Fragment E). Don’t pay attention to the Amino Acid sequence in the below captions as I didnt bother to check if they were in frame for the snap shot. The FCS is in frame above.

Other Neighboring BsaXI site has GTG and GTT overhangs- also not complementary to the other BsaXI sites.

So you can see that none of the other BsaXI sites in Fragment E have compatible overhangs so this digested product can only assemble one way after you replace any piece of it you desire. That means you can put this Matryoska doll back together after you swap out an SEB/FCS cassette.
So the TypeIIs site left in SARs-CoV-2 is a very well thought out indexing system that enables researchers to cut and past various fragments of the genome into ever smaller pieces they can easily paste back together again.
Given the intense research focus on FCS sites and SEB, one would expect any engineered design to have the capacity to index various aspects of the genome for modification. We do in fact see this in SARs-CoV-2. I find this BsaXI site an additional level of evidence that supports Bruttel et al.
This is laboratory derived.
Some critiques of using Restriction enzymes like this are that in modern times one can order DNA fragments synthesized from an oligo synthesis house like IDT. While this is true, Its expensive if you want to tinker and make lots of changes. having very cheap means to shuffle the genome and not have to go back to expensive 30kb synthesis is where you eventually end up. Even shops like IDT don’t attempt to synthesize the entire 30kb in one go. They take a 3kb hierarchical approach much like Ham Smith as PCR has its limitations amplifying more than 10kb.
Omicron Addendum
A productive twitter convo on the paper asked the question… What does Omicron look like?
This is BA.1 compared to SARs-CoV-2.
Notice the number of BsaI and BsmBI sites are identical. They are at slightly different coordinates because Omicron has insertions and deletions which offset the coordinate system.

Layer in the BsaXI sites.

Notice the BsaXI sites that flank the interesting SEB/FCS site(In Blue on Lower SARs-CoV-2 restriction map) are now missing!
Let’s have a closer look.

The BsaXI cut sites are highlighted in Purple. The BsaXI recognition sequence is in Orange. There is a Red SNP (CTCC→CTCA) in Omicron that alters a Proline to Histidine mutation. This is a very important mutation in SEB/FCS. There is another mutation highlighted in Grey that marks the N→K mutation in SEB/FCS. It looks to me that someone took Wuhan1 and replaced Fragment E’s SEB/FCS site by using BsaXI to excise a small cassette containing SEB/FCS and replaced it with an ablated version that may reduce the super-antigen characteristics of SEB and reduce cytokine storms. We know from BUhan-1 (Boston University GOF experiment), that swapping Omicron spike into Wuhan-1 led to a decrease in lethality in humanized mice. We don’t know if this experiment increased transmissibility or RO.
10/24/22- One critique of this post was that the BsaXI site doesn’t completely excise the FCS site as it leaves one R behind. But that R (Red arrow below) is conserved in most Coronaviruses. So the BsaXI site perfectly excises the region in Black (Y-QTQTNSPRR-A). The Y and A are part of the BsaXI insert overhang. If you want to change those, it can also be done as the TypeIIs nature of BsaXI affords the overhangs to change (Ns). However changing those overhangs will require an additional change in the overlaps of the neighboring fragments. More work but not impossible.
Another critique implied RpYNO6 could recombine to generate the BsaI/BsmBI/BsaXI fingerprint. The Top restriction map is RpYNO6. Bottom is SARs-CoV-2. The restriction sites that change are highlighted in yellow. Since the yellow site alternate between the two, this would require multiple short recombination events. Double lottery fallacy.
And just to emphasize the point that TypeIIs restriction sites can be used in either orientation, there is a great paper from Ralph Baric’s lab that uses both approaches in the same paper and on the same construct. The sequences are listed below. The NCBI links to the FASTAs are hyperlinked so you can check my work with SnapGene. It’s important to verify this with the exact sequences from the paper as the figures depicting this are confusing.

This image in the paper has caused confusion as to where the restriction sites go. But the sequence files in NCBI can be downloaded and you can see for yourself in SnapGene where the sites are.
Here is the restriction map from 15Kb across the spike protein to the end of the genome. This is region, Baric was modifying with a GFP and a GFP-nLuc.

You can see the Synthetic construct Baric made, he amplified from plasmids (primers not listed) and reconstructed in a manner that retains the restriction sites in the genome. The gel below demonstrates the banding patterns that emerge when you amplify with these fragments from their 7 storage plasmids.

Once Baric decides to put the GFPs in frame of a gene, he cant introduce a new TypeIIs without changing the protein, so he resorts to No-See-Ums approach for this piece. Top Restriction map is of SARs-CoV-2 and the Bottom Restriction map is of Barics Synthetic construct with GFP added. No new restriction sites despite the addition of GFP.

So a paper submitted in April 2020 from Ralph Baric’s lab constructs SAR-CoV-2 synthetically, very silimar to as Bruttel et al predict. If you are familiar with the complexity and scope of this paper, I’m very skeptical this started after the pandemic. There is at least a years worth of work in this paper submitted in April of 2020.

I have further annotated this his Syn SARs-CoV-2 genome with the restriction sites he lists in his figure. @Jbkinney noted that the Baric may have used a clever approach of putting the BsaI in the PCR primer tails on the fragments that contained BsmBI sites and used BsmBI PCR primer tails on the fragments that contained BsaI sites. This approach leaves endogenous TypeIIs sites in the genome and removes the PCR tails before assembly. This approach also fits with his sequence.
Hopefully this demonstrates that there are many ways to skin this cat synthetically and while Baric may have taken a PCR approach with leaving groups that retain internal restriction sites, there are multiple solutions to the problem and the BsaI and BsmBI sites are in routine use for constructing SARs-CoV-2 synthetic assemblies.
To further support this, the Arginine (R) amino acids within the BsaXI sites that flank SEB/FCS in spike are coded with human codons ‘CGG-CGG’. This was one of the earlier hints at human modification. There are also two Amino Acids that change from Wuhan-1 → Omicron in the SEB/FCS site. What does this codon bias mean. There are several codons for each Amino Acid and organisms have codon dialects. Human tend to use CGG for Arginine and SARs does not. There is great paper thats maps all of the SARs-CoV-2 codon preferences. You can measure which codons it over uses and which ones it rarely ever uses.
In making the mutation seen in P→H and N→K from Wuhan-1 to Omicron you can see there is migration toward more rare codons. One of these mutations is C→A transversion (P→H). Transversions are rare in Coronaviruses. They tend to mutate as transitions. Transversions are when you mutate a base and it moves from a Purine to a Pyrimidine (A and G are Purines and are bigger bases, C and T are Pyrimidines). Changing a base such that it stays in the chemical family (transitions) is favored over changing bases that switch chemical families (transversions).
So mutations that occur in Omicron’s SEB/FCS site are significant changes. When N’s are removed, Glycosylation is altered. When prolines change in the FCS site, ribosomal pausing changes (Postnikova et al) and likely the activity of the FCS is attenuated. These critical changes (amongst many others) are all addressable with a cloning schema described by Ralph Baric in April of 2020. The question still remains, if all of the other mutations in Omicron are easily addressed with such a design?
This data does not implicate the Baric lab but it is a reminder that the recipe to make SARs-CoV-2 as described by Bruttel et al was submitted for publication in April of 2020 and available for all to replicate by summer 2020. Given the weight of the paper and the immense amount of work it describes, I find it hard to believe that work all initiated post pandemic. If it was submitted in April, it was being worked on before the pandemic emerged.
This does leave one wondering why Congress hasn’t subpoena’d the Oligo Synthesis shops (IDT) in their jurisdiction to see if UNC, RML or any of the labs working on GOF (BU included now) have order oligos that would support such a design and when?
Best thing they could do is Subpeona the oligo synthesis shops for oligos ordered in 2019.
BLAST these against SV2.
That would lead to the perp
A frikkin' home run read... made my night...