Chakraborty Open Review
Why SRA sleuthing is so valuable

Many modRNA vaccine enthusiasts are predictably complaining that Chakraborty isn’t peer reviewed. This is a bit lazy. When a paper emerges that points to already peer reviewed data and simply claims there is other stuff in there, all anyone has to do is download the data and check for themselves.
It’s like a Sudoku puzzle. Hard to solve but easy to verify.
Chakraborty did the hard part of scrutinizing the Ryan et al papers reads. Now all anyone has to do is the easy part and check if those reads are really there.
Yup.. They are!
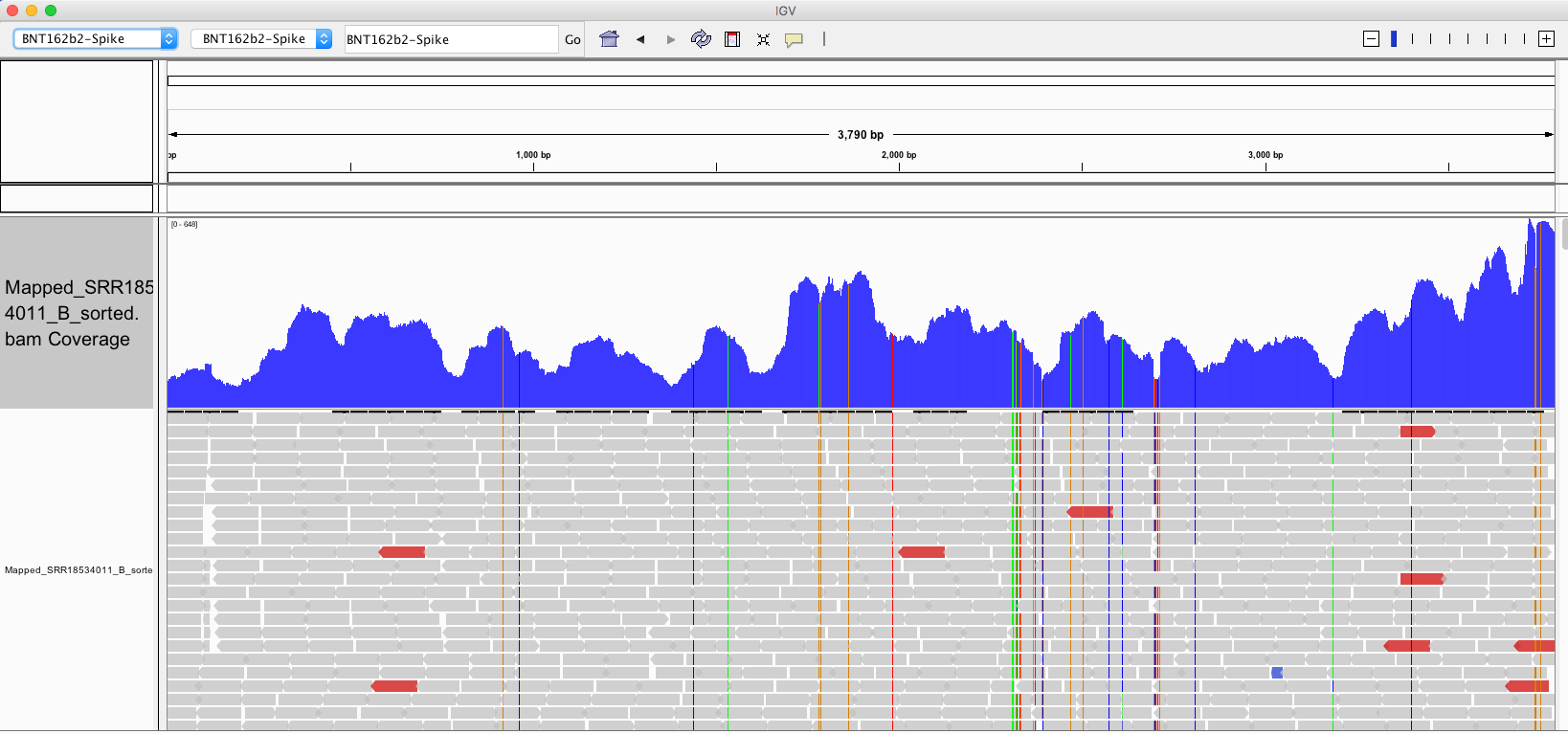
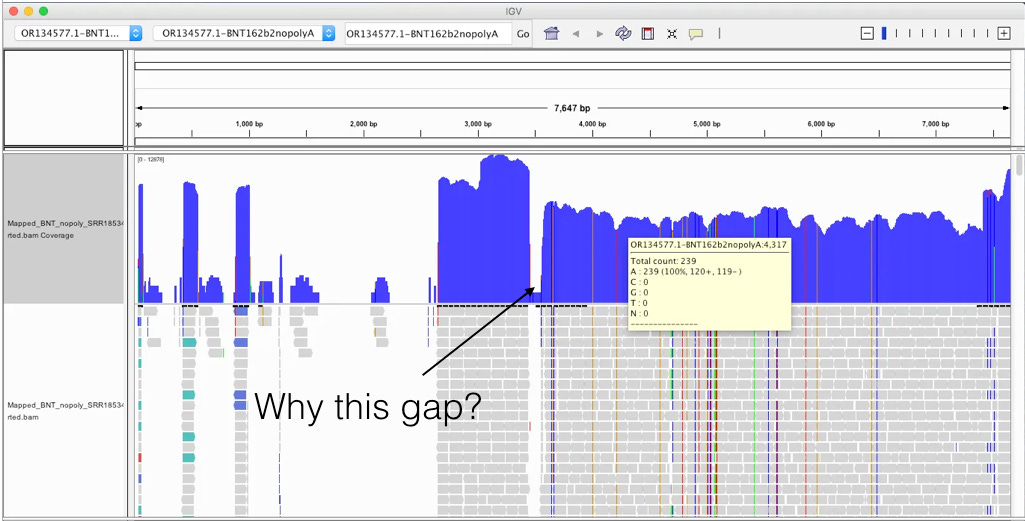
The above picture is an IGV coverage plot of the reads mapped to BNT162b2 spike. Each horizontal grey arrow is a read. Vertical lines are SNPs or Omicron differences. Our reference is a collage of omicron & Wuhan-1 as we sequenced a bivalent vaccine to make the reference. So when we sequence a monovalent vaccine the tools highlight all the differences between the reads and the reference. The top Blue histogram is the read depth across the spike. The highest part on the chart is ~600X coverage but the average over spike is closer to ~250X coverage (I’m eyeballing). This lines up with what Chakraborty published for this sample.
For those interested in how to do this yourself, there is great news. ChatGPT has made it much easier for people without a PhD in bioinformatics to do this.
I’m going to show you how.
If you are not interested in learning how to do this, here is TL/DR summary…but you need to read the conclusions as there is a whopper in here.
The first sample in Chakraborty’s Australian study is SRR1854011 and I have confirmed there is BNT162b2 sequence that could only come from the DNA not the RNA in a sample that shouldn’t have any.
These samples were collected with Qiagen RNA Blood preps (see conclusions on the confusion over this). These use columns that select against the capture of DNA and optionally recommend the users DNaseI the sample. Despite this bias against DNA, its still showing up. And its in patients blood stream and contains SV40 promoters. We should expect some spike sequence as they were vaccinated and that RNA can get turned into DNA in the DNA sequencing process. Library construction kits that target RNA convert them into DNA first as sequencers (other than ONT) can only read DNA. We shouldn’t see the plasmid backbone or the vector sequence at all. But there is catch that you will have to read in the conclusions.
TUTORIAL
The Tables in Chakraborty have NCBI-SRA accession numbers like SRR18534011. The SRA is the Short Read Archive. When you publish genomics papers, it’s a sin to not put your data in here or a similar repository. This sin was committed by Stanford when they sequenced the vaccine first (in GitHub) but never put the raw reads public despite multiple requests for the data. They hid the problem in their desire to limit vax hesitancy and history will not look kindly on this elitist behavior.
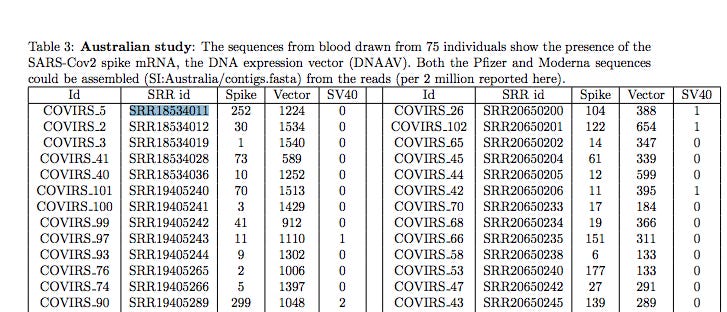
If you go to the NCBI SRA website, you can look up the project SRR18534011.
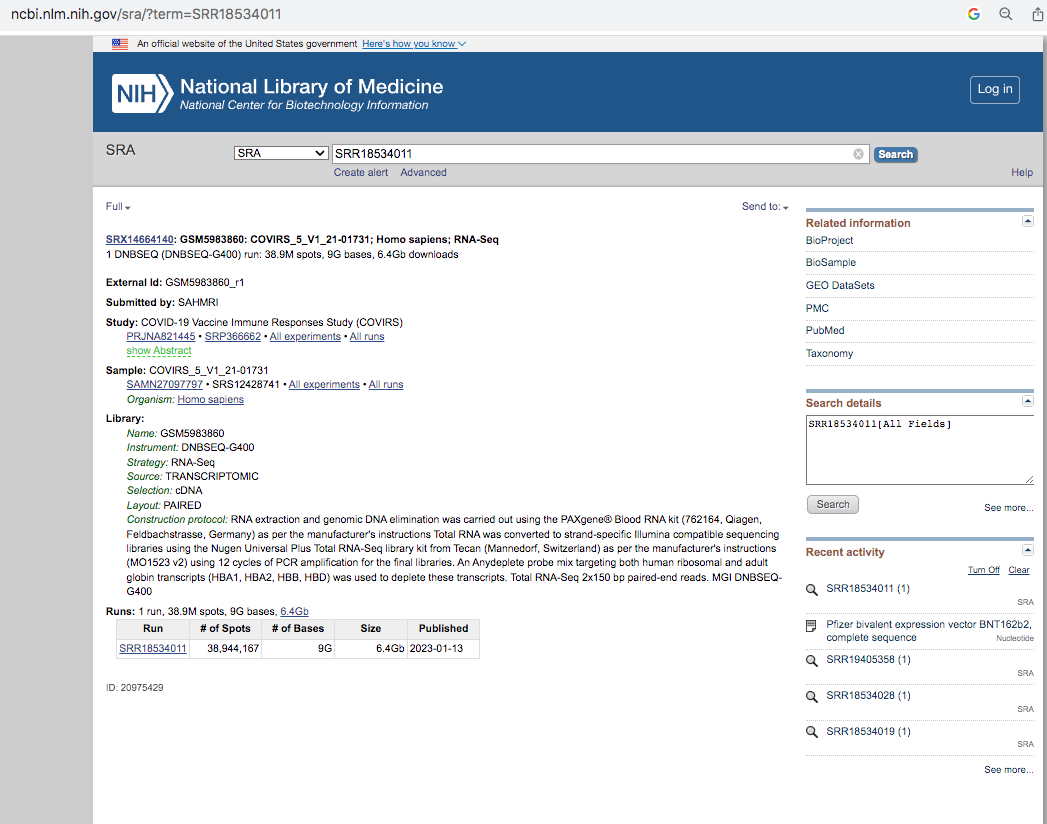
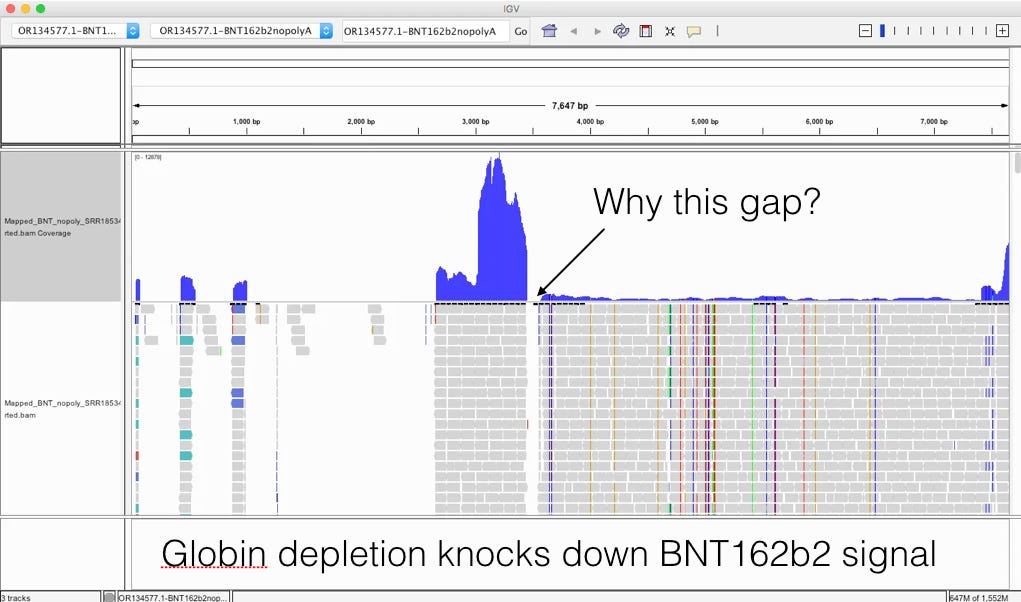
It’s important to read the methods section as it gives you some glimpse as to how this RNA was processed. 7 Days after dose 1. Note there was a globin depletion step!
When sequencing RNA, high copy transcripts often consume all the sequencing reads so people sometimes like to hybridize them out to get a cheaper picture of the non-globin transcripts. This is serious flaw in this study as the BNT162b2 sequence has a globin sequence and this depletion step could remove the very sequences they are trying to track. Post-script. I emailed the authors and tracking the residual RNA wasn’t am aim of their study and globin depletion is quite common when handling blood samples. We really need studies designed to find this.
This likely explains the gap in sequence coverage we are going to see below.

Since the files are large and this is government run, you can’t just download the data like its a Rumble video. You have to install custom software and perform some DMV like rituals to get the data. I used to joke that this was intentional to keep the public from asking too many questions and to ensure only skilled bioinformatics people are combing through the underwear as most of them won’t bite the holy NIH hand (tinfoil sarcasm).
A single patient sequencing file for this project is 6-10Gb and there are 75 patients. I’m not going to download them all as that will kill my drive space.
But downloading the first one on the list and mapping all the reads against the PBiv reference we have in NCBI shows ~500X coverage over the plasmids! That is remarkable.
You will need familiarity with MacOSx or Linux command lines. The first tool you will need is the SRA-Toolkit. Since their website is clunky, I would just ask ChatGPT how to install the sra-toolkit with brew or similar command line tools.
Warning-This is a several hour process. The most frustrating aspect of it will be downloading and installing the various tools. But once you have those installed it makes it very easy for you to repeat this on future genomes of interest.
You will need
sra-toolkit, cutadapt, bwa, samtools and IGV.
Thankfully ChatGPT makes it easy to download each one of these and trouble shoot any version issues with your Mac.
If the version number comes back clean you have a clean install.
If this sra install gives you problems you can download the trimmed read data from these Mega links (imagine if the SRA were as simple as Mega). They are ~4Gb each.
Once you have reads you need to trim the sequencing adaptors off. If you download the MEGA above link you can skip this step.
Trimming requires Cutadapt. ChatGPT can also lead you through how to install this. It can also tell you which primer sequences you need to feed the Cutadapt command line for trimming and write the command line for you!
This will take an ~hour on a single core as the program needs to scan every read for these sequences and clip them off and rewrite the files.
Once you have trimmed reads the next step is mapping those reads.
We use bwa mem to map reads to a reference. ChatGPT can help you install this as well. You will also need samtools installed. The final install of software is IGV but that can be downloaded through a web browser (no command lines).
First you must index the genome with
bwa index fastafile.fa
The fastafile.fa needs to point to the Pfizer vaccine sequence I have a link to below.
Then ChatGPT can write you a command line to map the two trimmed read files against your genome. You may need to pull the CPU threads back. I have a very powerful 96Gb RAM MacBook Pro that can handle 96 threads on its CPUs. -t 24 may be more reasonable for a laptop.
When you are done with this… you probably want to screen this BAM file for only mapped reads. The reads that don’t match your reference are human and just bloat your BAM file. I have also trimmed the polyA sequences out of the Pfizer vaccine sequence as those recruit human reads.
This will significantly shrink the size of the BAM file.
Now you must index it before you can view it in IGV
samtools index Bam_File.bam
Insert the name of your bam file in BAM_File.bam
Now you are ready to load 2 files into an IGV browser. Open up IGV and use the Genome tab to load the Pfizer reference above. Then use the File menu to open the BAM file.
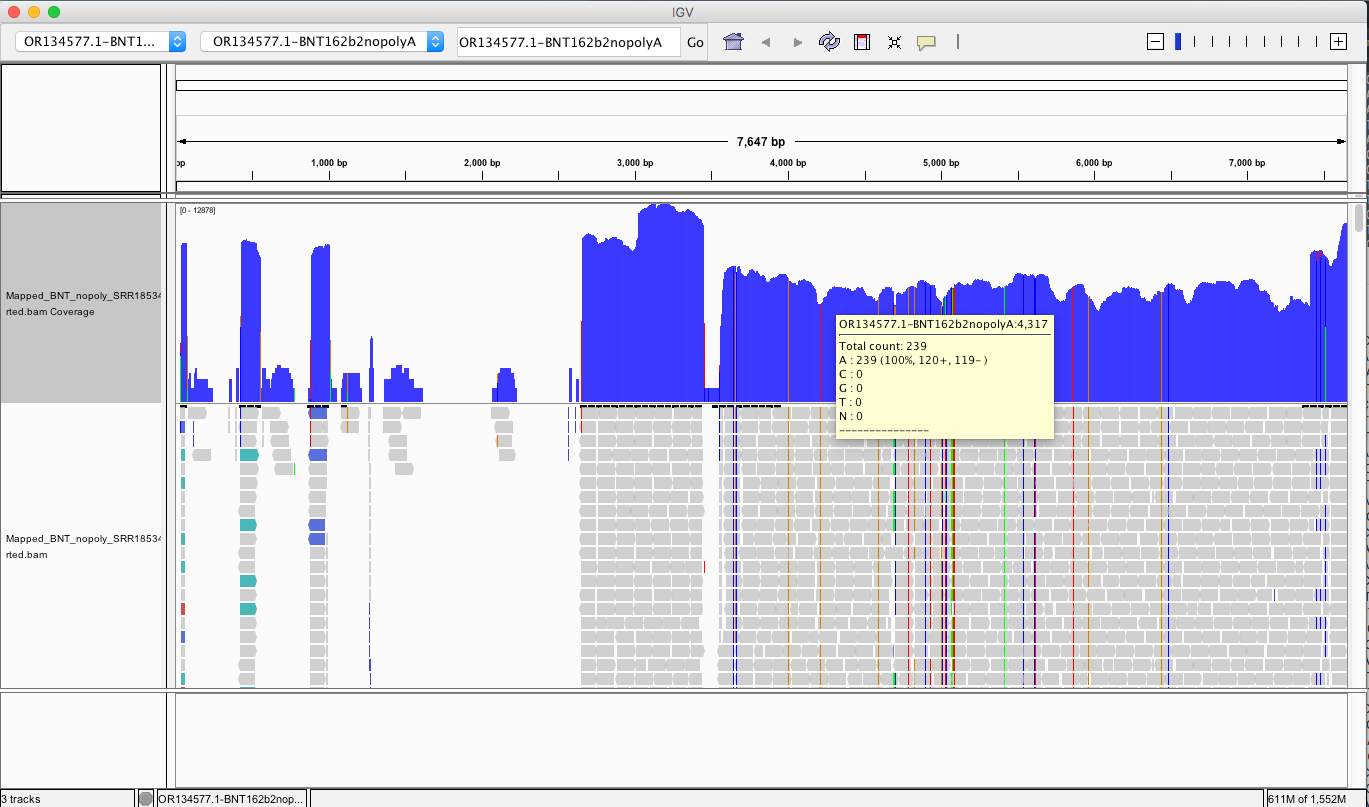
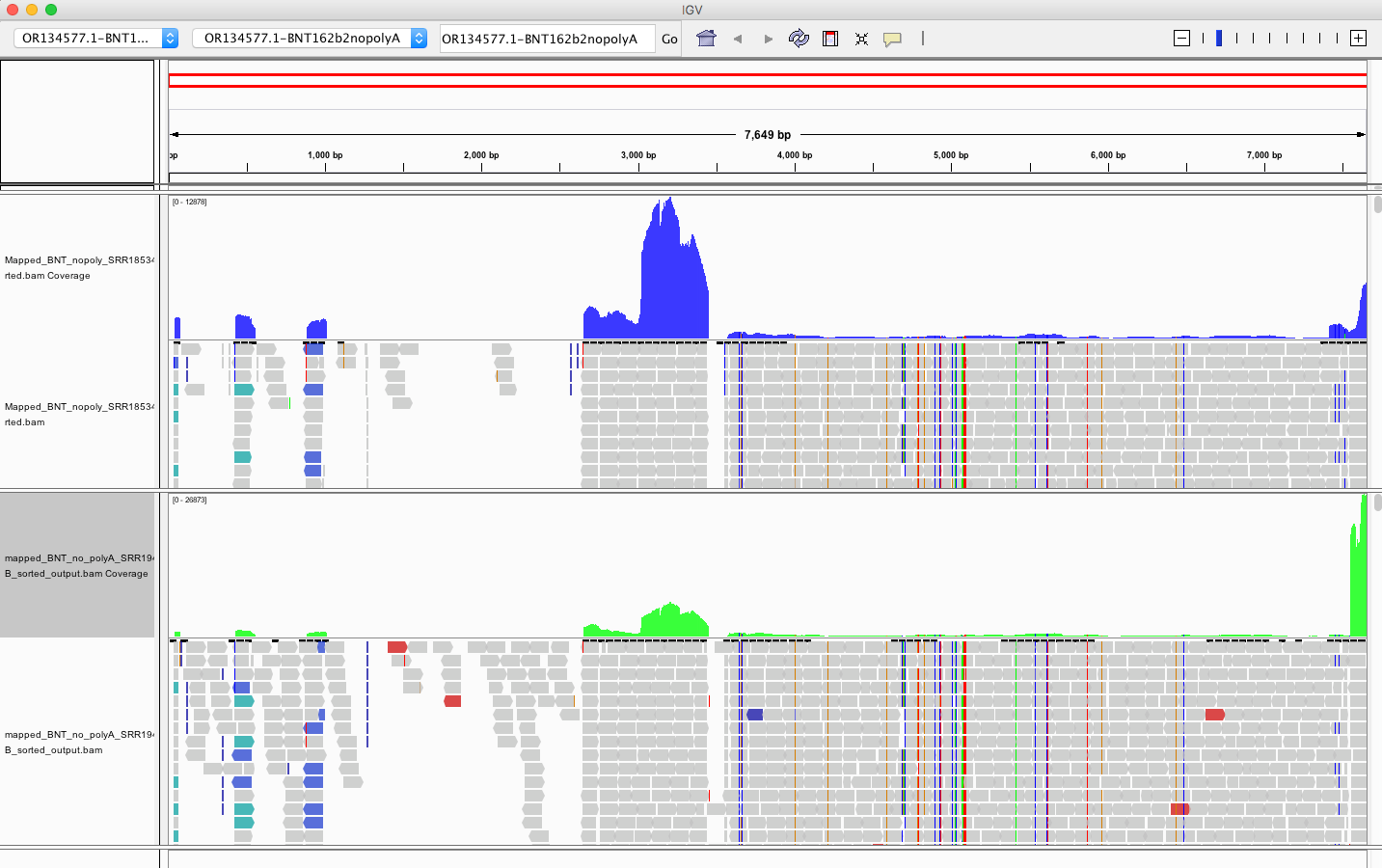
My IGV defaults to viewing the coverage in LOG scale which can be deceiving. Below is linear scale.
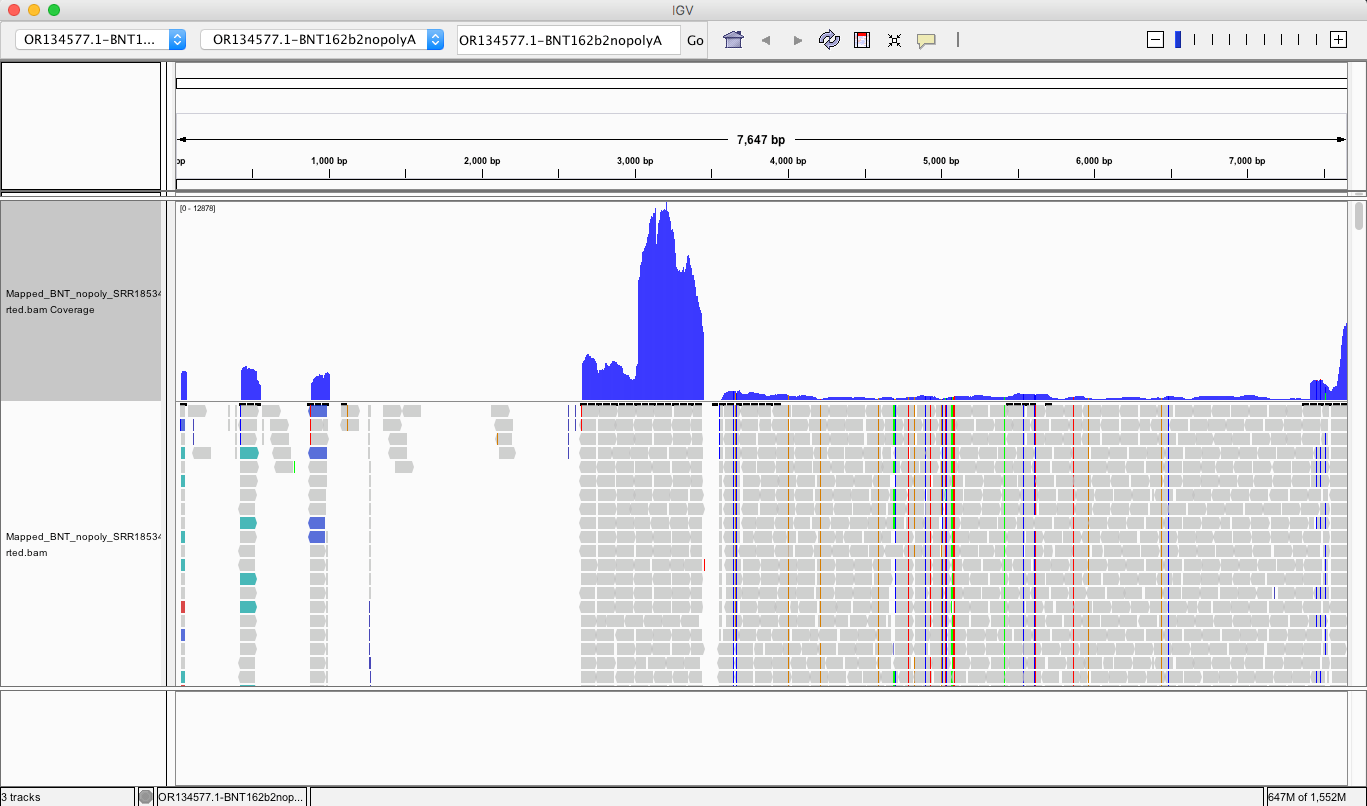
The region with the highest coverage (middle) is the Bacterial origin of replication. RNA-seq methods will capture RNA and ,in turn, capture background DNA. The spike region is unique to the vaccine and you can see its coverage is much lower (Right side with SNPs). Other parts of the vector are often digested more readily with DNaseI. If these are smaller pieces of DNA, the Qiagen Preps likely didn’t capture them. So you should expect more gaps in the parts of the plasmid we know digest more readily with DNaseI (not spike). Spike doesn’t digest well with DNaseI as it has complementary RNA that forms RNA:DNA hybrids and DNaseI doesnt operate on these molecules (Sutton et al). This suppresses the digestion 100 fold over spike. Hence the vaccines are contaminated with 100X more of Spike DNA than vector backbone DNA (very lot dependent and more so in Moderna). This is why the regulators can’t see this. They are only measuring the Kan gene (in the vector backbone) because Pharma told them to look there with PCR. Nice trick they played.
The SV40 regions do slip through at lower copy number than spike. Some of these reads are short alignments with softclips that should be scrutinized for genome integration events or other artifacts.
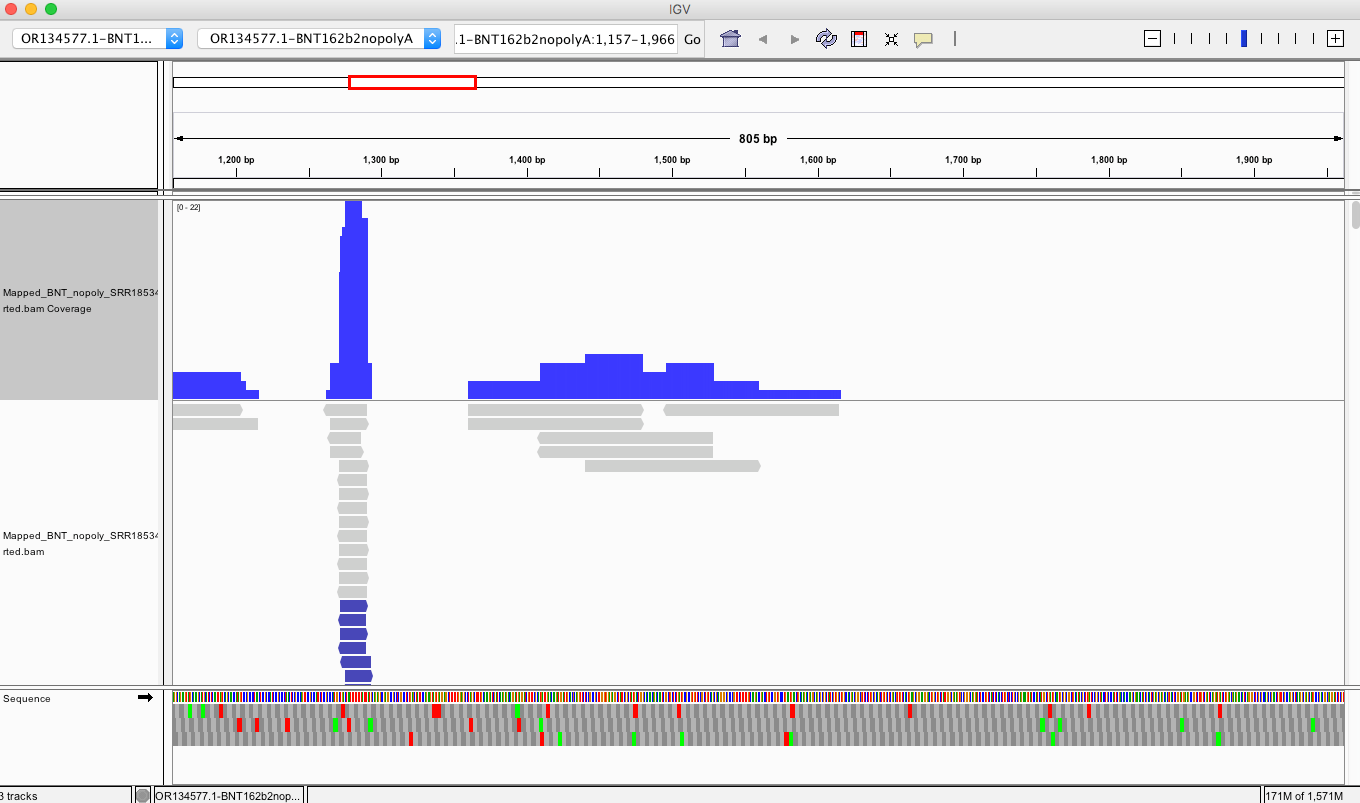
The vector regions (Left) shouldn’t express RNA (in blood) that gets pulled into RNA preps but their DNA will be captured to the extent the RNA preps allowed for it. The spike region (Right with SNPs) has RNA delivered in the vaccine so some of the higher coverage spike region may be RNA that is converted into DNA in the library construction process which prepares these samples for sequencing on MGI platforms.
It really comes down to if the people who originally processed these samples used a DNaseI step. If they did, then these spike sequences are likely cDNA conversions of the vaccine RNA. But the vector sequence (Bacterial Ori, F1 Ori, SV40 etc) shouldn’t be there at all and shouldn’t be making RNA. Blood is usually clean of bacteria unless the patients are septic. These are most likely DNA remnants from the vaccine that are partially captured by their RNA prep methods.
One artifact we have to look out for is index hopping. Since they are serial SRA numbers its very likely all of these RNA samples were DNA barcoded, pooled and run on the same MGI chip. Index hopping occurs when one DNA barcode gets misread as another and thus the de-pooling of the data is slightly compromised. This usually occurs at less then 1 read per 10K-100K on the DNBSEQ-G400. It can be 1-2% on Illumina.

MGI has much lower index hopping than Illumina so the frequency is very low but this might explain why Chakraborty is finding SV40 sequences in the Moderna samples?
Were the samples run on the same MGI Chip? You can usually discern this by cracking open the Fastq files and looking at the read names. The chip ID is usually in the read name.
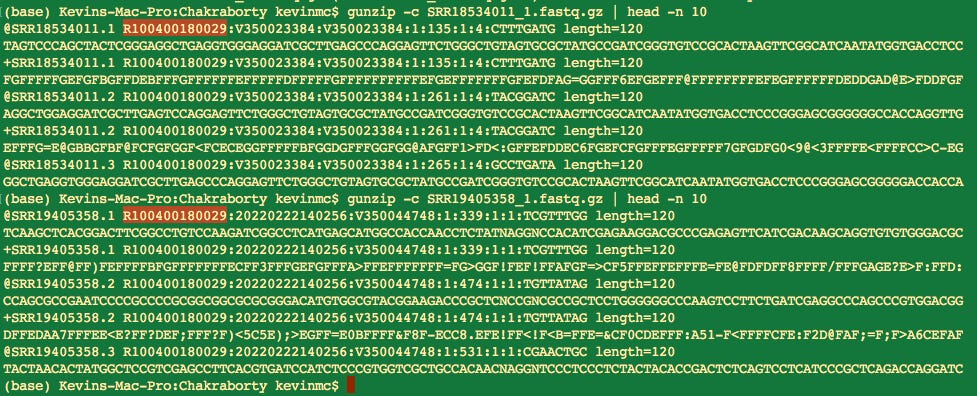
Indeed, these samples were all run on Chip R100400180029 so the SV40 findings in Moderna I would still interpret with some caution.
Conclusions-
The globin depletion step in Ryan et al is a serious flaw for our purposes of looking for nucleic acids in the blood (This wasnt an aim of their study- personal communication with the communicating author). Not Chakraborty’s problem but a problem from the original study.
Looking at this in non-LOG scale….
If you look at the soft clips of those reads they map to HBA1 and HBA2? These are globin genes they admit to depleting. Soft clipped reads are reads that map well for one length of the read but then diverge. In the yellow box below you can see Left 87bp Soft Clip. Thats more than half the read that hangs off.
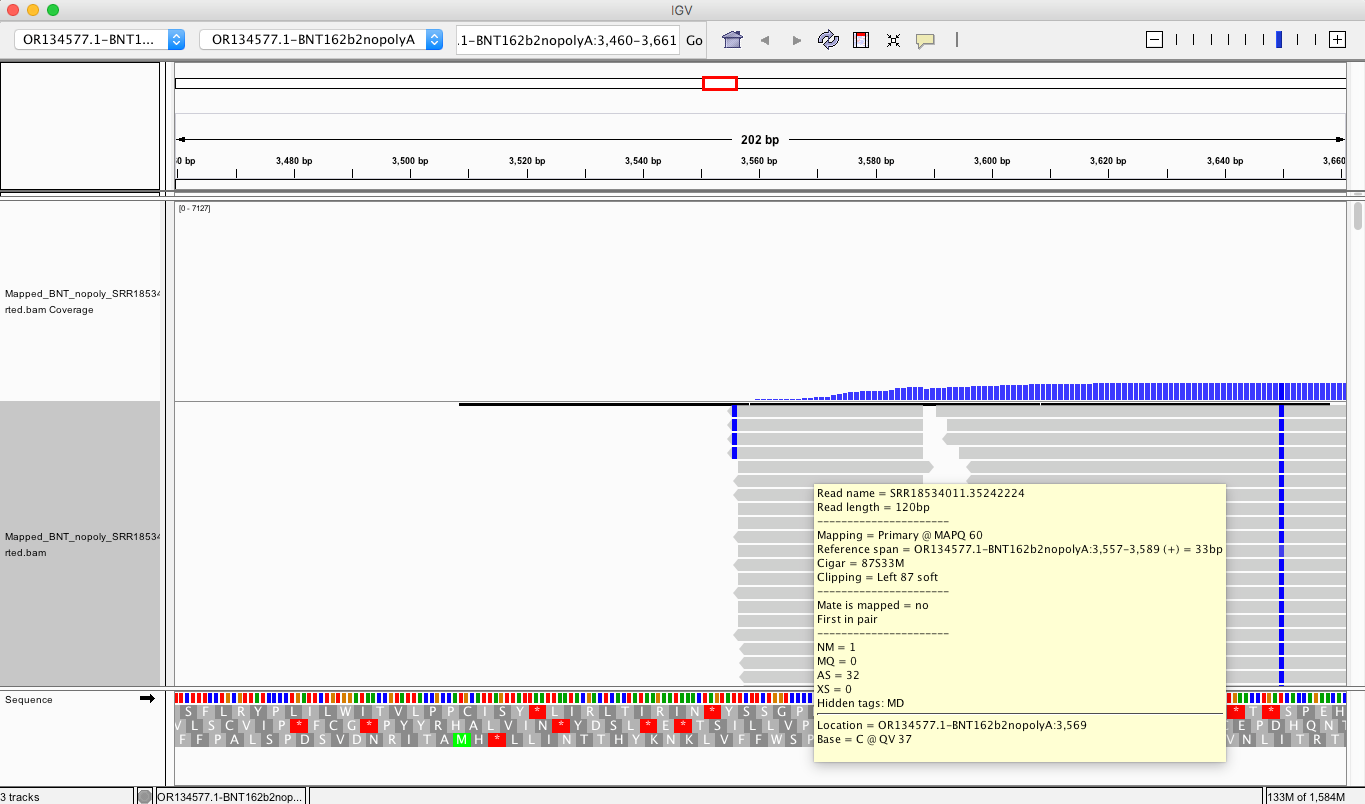
If you BLAT those reads they map to the Globin genes they claim to deplete and they map to BNT162b2! That tells us their depletion probes can deplete BNT162b2 and suppress the RNA/DNA persistence data in the paper. This pattern repeats itself in other samples (SRR1940538).
BLAT of soft clipped reads below. Just take the entire read sequence and put it into BLAT and you’ll see the entire read maps to HBA genes. These are reads that also map to BNT162b2. So if they are depleting this gene they are also depleting the mRNA transcript for BNT162b2.


In other words they BLAT to the very regions of the genome that their methods claim they are trying to deplete while part of the reads map to the BNT162b2 transcript. This demonstrates that the depletion probes they are using have homology to BNT162b2. If one is trying to monitor RNA persistence (They were not aiming to do this), the use this depletion method is going to suppress the signal for capturing the modRNA.
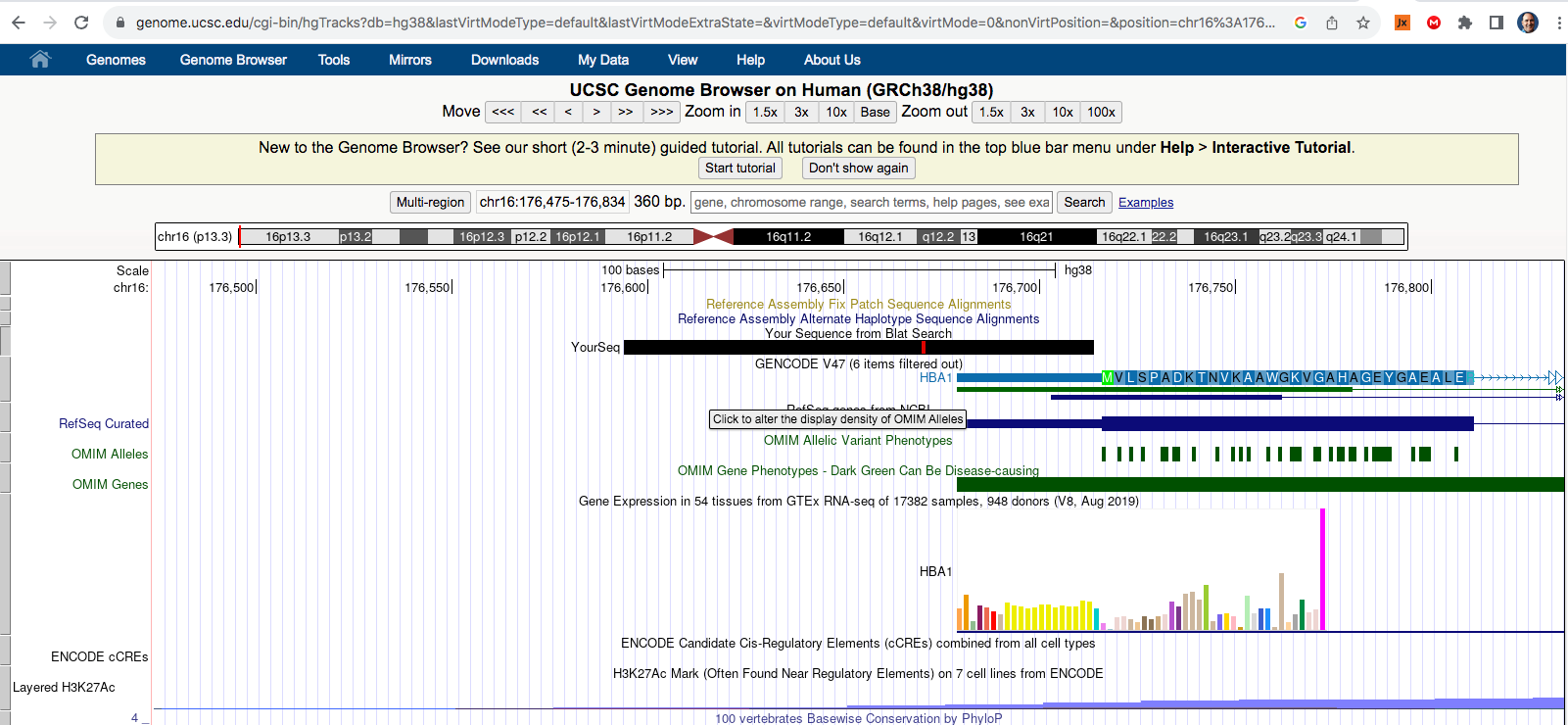
This likely explains why Ryan et al didn’t see vax RNA at 28 days (V2B below). They are depleting it from the picture with their protocol. This would explain why the vector DNA is so much higher coverage than the spike.
After speaking to the authors, the RNAseq purple dots in this figure 1A do not represent positivity for the vaccine but just that those experiments were run at those times.
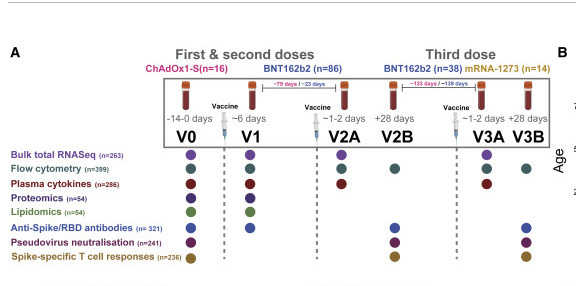
Its a bit harder to go much deeper on this without communication with the Ryan et al authors. One point of confusion is that their methods in the paper don’t match those in NCBI. NCBI methods claim a Qiagen kit was used for the RNA. The methods from the paper below state RiboPure methods from Invitrogen so its unclear if a DNaseI was used.
Post script- After contacting the authors they responding thank us for pointing out the differences and that the manuscript has the correct methods and they correcting NCBI methods. They did use a DNA depletion step which means any DNA signal we can find is heavily suppressed.
Both methods point to globin depletion protocols being used. Their data shows sequence evidence for depletion probe homology to BNT162b2 5’UTR. This could deplete the whole transcript and suppress spike signal.

How do I know these reads are from RNA or DNA in the sequence? They used a stranded RNA kit which should only capture Crick Stranded RNA. If you capture any Watson stranded molecules those could only come from the DNA. You can easily count the Watson vs Crick ratio of stranded RNA-seq data with samtools view -F 16 vs -f 16.
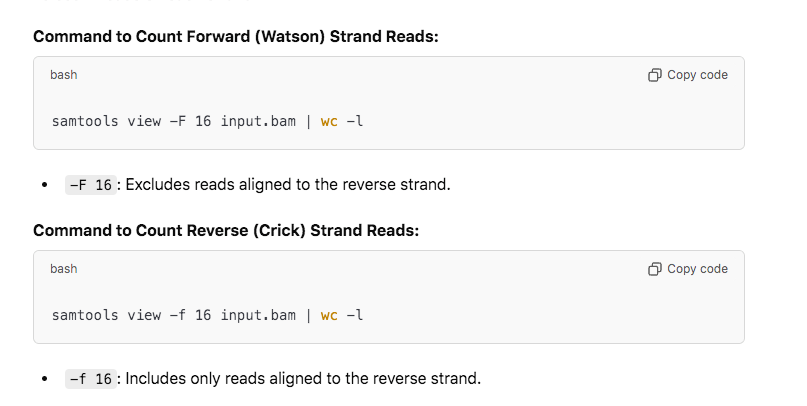

We have equal amounts of reads mapping to both strands. This is spike DNA.
Most peer reviews will never go this deep.
In summary, the vector sequences shouldn’t be showing up. The equal number of Watson vs Crick strands shouldn’t be showing up. The total signal is likely being suppressed in the Ryan et al study as the RNA preps don’t capture DNA efficiently, likely loose small molecules with Ethanol washes and their depletion method interacts with BNT162b2. I hope this gives you some confidence in the signal that what Chakraborty is seeing can’t be easily dismissed and the signal would likely be even higher if different methods (designed to capture the small DNA) were deployed.
Appendix-
The 5’UTR in BNT162b2 is a beta globin sequence from HBA1 and HBA2. These are the genes Ryan et al claim to have depleted. Therefor their RNA persistence data is suppressed.
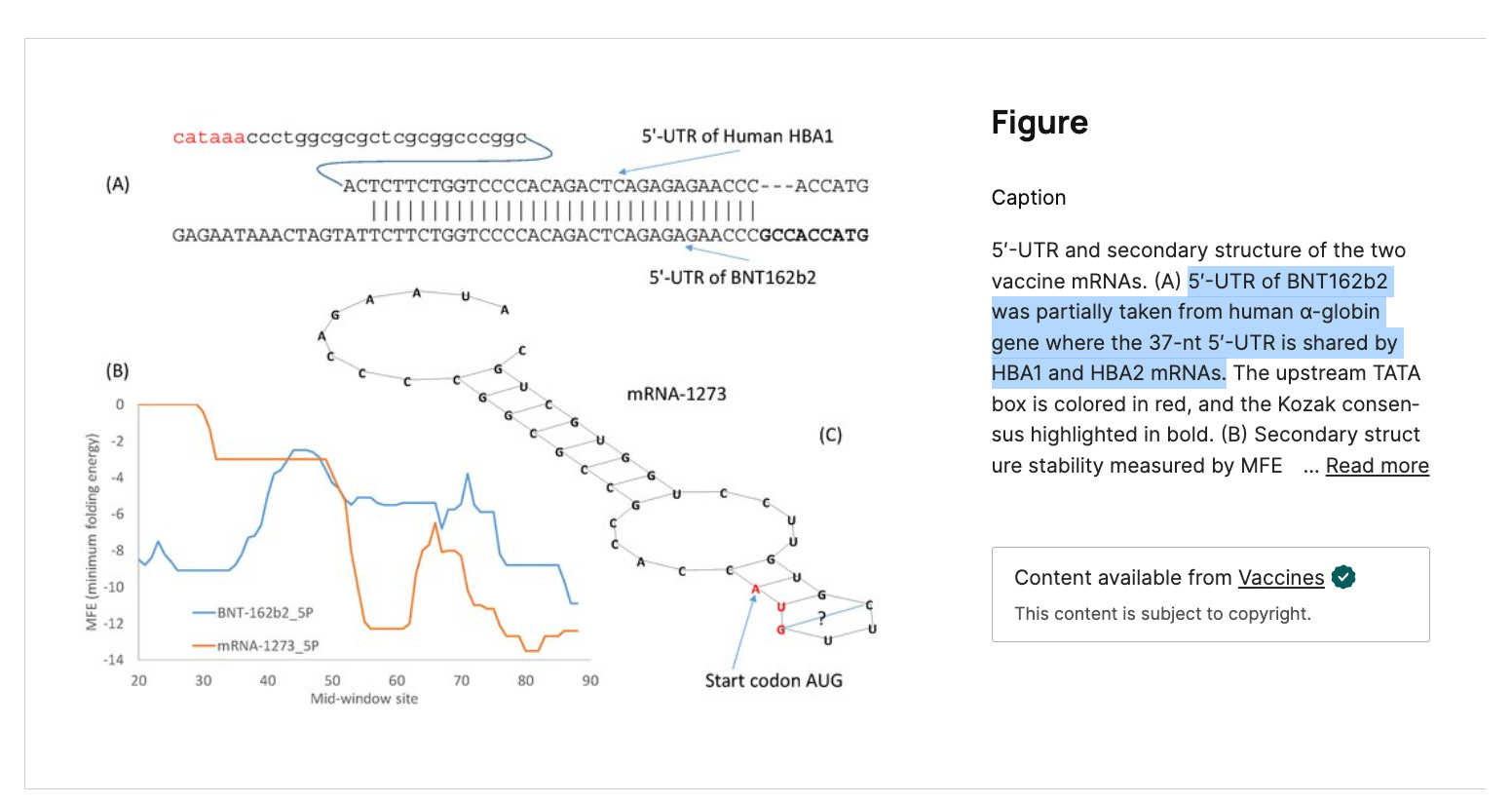
Here is the 5’UTR beta globin sequence in the Pfizer plasmid in blue.

Post Script-
Phillip Buckhaults also downloaded all of these samples and all of them have these odd peaks over the Bacterial origin of replication and the F1 origin of replication suggesting there is contributing DNA from a plasmid used to make one of their reagents. Some of the samples have more uniform coverage over the plasmid backbone (SRR20650192 & SRR19405260). These samples may be better candidates for whole genome assembly.
Peer Review? You mean offer a few Scientists a $100 million research grant to cover up the facts and save billions of dollars in lawsuits? After the last few years I’ve come to realise that peer reviewers are the HR department of Big Pharma
Thank you for the detailed instructions! It's been over a decade since I did any bioinformatics work, and needed a refresher.