

Chocolate in my Purrnutbutter
The mysterious case of a Spike Protein encoded Plasmid


This week I was alerted to a very interesting Twitter thread that showcased a BioProject in NCBI that contained sequenced Pseudomonas aeruginosa genomes collected from patients in China. P.aeruginosa is an organism that often infects Cystic Fibrosis patients lungs. Since cystic fibrosis patients often seek out medical cannabis licenses, some jurisdictions demand P.aeruginosa qPCR testing for cannabis flowers. As a result, our team at Medicinal Genomics have sequenced a lot of P.aeruginosa genomes and we’ve never found a plasmid with a spike protein in them.

The article that stimulated a great Twitter discussion on this spike protein plasmid is shown below. The conversation is much more cordial when the threat of censorship is lifted from the participants.
The collection date in NCBI is 2019 but the submission dates into NCBI are 2022?

From the article posted in the twitter thread above.

Given the large gap in collection time to NCBI submission and the fact that this time interval traverses the Zoonosis theory born on date of SARs-CoV-2 in Dec19 , this sequence becomes pivotal to the origin debate. The spike protein is 100% identical to the amino acid sequence of C19 spike minus the 4 critical A.As in the FCS. There are some mods on the 5’ end and the 3’ end of the spike protein that are interesting. The nucleotide sequence is 73% identical to the virus and 88% identical to BNT162b2 and mRNA-1273. This implies codon optimization occurred and there is no question that this spike is man made as its in a commonly used E.coli/Mammalian expression vector.
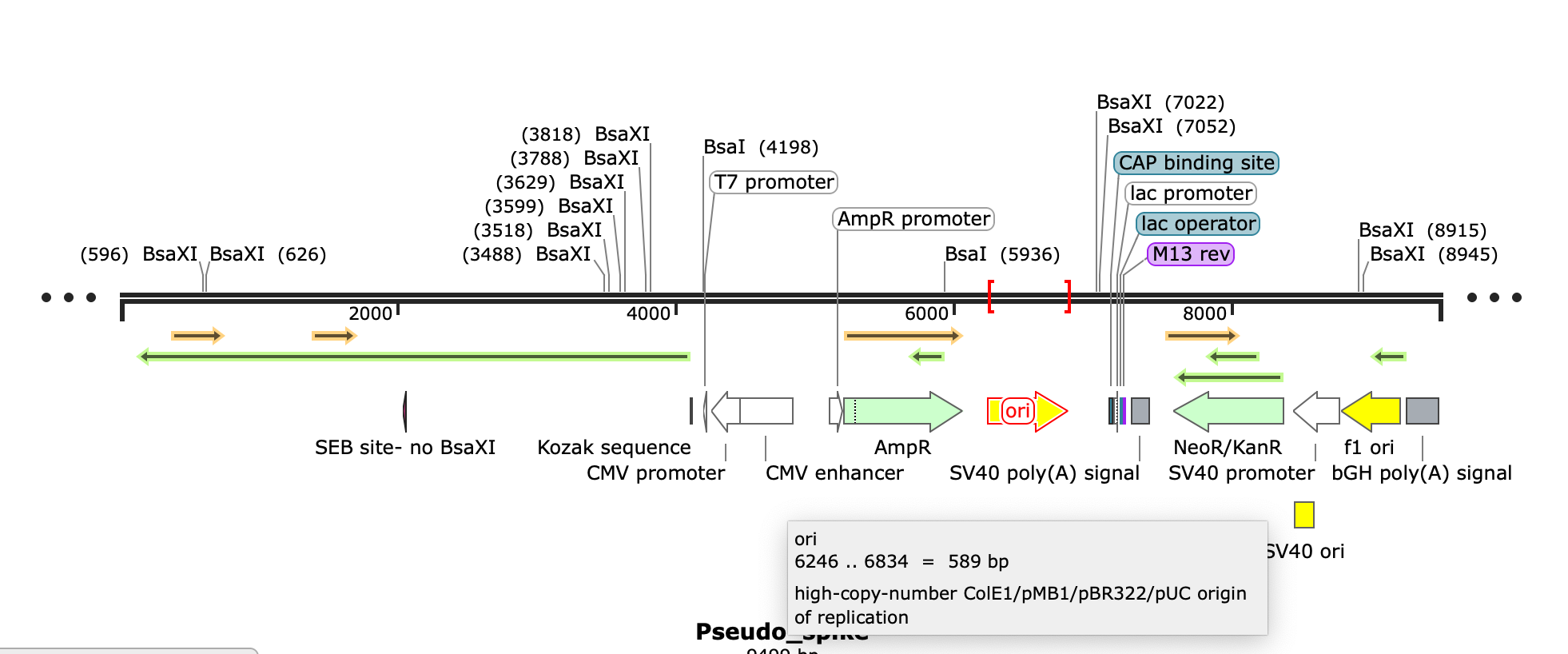
This expression vector displays the intent of the engineer. It has 3 selectable markers used for bacteria and mammalian cells.
Selectable markers are very important to maintain populations of plasmid transformed cells. If you grow E.coli in the presence of ampicillin, the only cells that survive are the cells which were transformed with your plasmid containing the antibiotic resistance gene. If you don’t have a selectable marker, your plasmid dilutes with each 30 minute doubling time of E.coli.
So for this plasmid to be stably replicating in a P.aeruginosa strain collected from a patient, there has to be some selection to maintain that plasmid in-situ (in patient) or in-vivo (cell culture) cultivation of the P.aeruginosa isolate.
Let’s look at the selectable markers and see if they make sense?
The plasmid has the AmpR gene often used for Ampicillin resistance when cloning into E.coli. It has a lac promoter with M13 primer sites complete with a high copy plasmid origin of replication known as ColE1. We used ColE1 vectors for part of the human genome project. The pUC origin listed below is known to make 500 copies of plasmid in every E.coli cell so a 500:1 copy number of plasmid to E.coli genome equivalents. Early versions of ColE1 made 25-30 copies per genome. Turbo charged version of the origin found in pUC are 500-700 copies. Given this is a 9kb plasmid, let’s assume the lower copy number ColE1 estimates (25-30 copies/cell).
This copy number detail is critical and will emerge later as one of the most important threads to pull on.
The problem with AmpR as a selectable marker is that this Pseudomonas strain (and many just like it) are already ampicillin resistant with a chromosomal copy of the gene.




The other two markers Kan and Neo are also notoriously poor choices for P.aeruginosa culturing. It might be helpful to sleuth this particular genome for divergances from what commonly infects patients. I stopped genome sleuthing once I saw these papers as the copy number issue caught my eye.

While the BioProject in NCBI has no raw reads in the SRA for analysis (if anyone finds them , please forward), it does have some assembly statistics which give you the depth of coverage of each contig. The contig with the spike protein in the ColE1 plasmid is only 4.2X coverage and these genomes are at 30X coverage? The blue contig below is the spike protein encoded plasmid. So the coverage of the ColE1 plasmid is 2-3 orders of magnitude off. Being a high copy plasmid it should have 20-30X more coverage than the 30X genome coverage. Instead it has only 1/7th of the coverage of the genome.


This tells us that this plasmid was not transformed into P.aeruginosa and as a result we cannot assume it existed at the collection date in 2019. Its is more likely that it was a contaminant in the sequencing process (too high plasmid coverage to be index hopping on the NovaSeq) and we don’t know when the sample was actually sequenced. If it was sequenced before Dec 2019, then the Zoonosis hypothesis just met its grim reaper.

If the sequencing date was after the publication of the Spike protein from C19, then it’s not that surprising that a sequencing lab may have ‘spiked’ some chocolate in the peanut-butter. Often times there is meta information in the raw sequencing read files (Fastq files) that can point to the flow cell barcode numbers which will have a known manufacturing date. This doesn’t provide perfect temporal resolution as there are flowcell shipment delays and W.I.P (Work in Process) delays in sequencing factories. In addition, these text files can be easily manipulated with a command line perl script.
So how do we implement immutable time stamps into high risk research like virology?
The solution to this sequencing data forensics problem is something Medicinal Genomics pioneered in the cannabis sequencing space. Due to the unique intellectual property laws in federally illegal markets, you cannot trademark cannabis but you can patent it. Legality is not a requirement for plant patents (yes, plants that are crafted by the hand of wo/man remain patent eligible). The notary system is federal so getting an ironclad born on date (without the use of federal notaries that object to cannabis) requires one notarize your DNA sequencing on immutable distributed ledgers like Bitcoin.
Blockchains are the least efficient means to store large datasets, so instead, a hash of the sequencing files are spent into the OP_RETURN of a time-stamped Bitcoin transaction using a service Like OpenTime Stamps. This provides an immutable time stamp of the born on date of the sequencing data. If one letter changes in that fastq file, the entire hash changes and won’t match the hash you etched into the blockchain.
GOF work may be restricted again given what just happened. There will also be a grey area of research where labs working on curing a disease accidentally make it worse.
This unforeseen nearGOF research needs a framework for responsible stewardship of data so we can learn from leaks and fix them.
Labs working on nearGOF should be required to etch their fastq files into Bitcoin so proper forensics can be performed.
Kannapedia.net hosts over 1700 Cannabis genomes using this ‘Hash and Stash’ method of data management. Most clients utilize this to prove their strain existed at a certain date so they aren’t exposed to submarine plant patents. The data has been used in court to resolve a dispute over stolen genetics.
We have built a similar public resource for Psilocybe cubensis at Psilocydia.net. This large public repository of medically valuable organisms ensures the prior art remains provably in the public domain.
If you want to defang the Pharma-regulatory fascism being exerted on the healthcare system today we need to cement the ‘OTC’ medicinal organisms into the public domain to thwart off patent thickets and other regulatory capture games. Privatization of natures pharmacy while socializing the risks through liability waivers and mandates is not the future of medicine. To decentralize medicine, we must advance the science behind these valuable natural pharmacies and open access those discoveries as best we can.
Many assume drugs that have been used for centuries can’t be patented. I beg you to review what the FDA did to Cannabidiol. It has made the case that CBD was never a medicine before this century and awarded an IND (investigational new drug) to GW Pharma. It’s therefore no longer a supplement and dispensaries can’t have medical use information on their labels.
Building ironclad prior art starts with transparency and high fidelity immutable data stewardship.
Something the field of virology should emulate given the lack of public faith in their data stewardship since 2020.
A healthy debate has ensued over this on Twitter.
One thing that was puzzling is that the 4 plasmids were all different? This led some to believe it cant be a contamination as that would require 4 different contaminations. On the flip side of this argument… No one sends one plasmid for sequencing on a NovaSeq. The output of the machine is like using Niagra falls to put out a match. People send libraries of plasmids for sequencing but how did they manage to get only one plasmid per genome? This could be explained by poor tip washing on a 96 well robot. It handles a plate clones making one library and the next plate it touches and contaminates in a very well specific pattern.

The other sign of contamination is the bi-modal coverage maps. We have more than 1 contig at log scale lower coverage. We have a whole collection of them and I’m curious what they all BLAST to and how many of them are out of place for a P.aeruginosa genome.

I did scan a few of these outlier coverage contigs and noticed many are hits to other pseudomonas strains.
Oddly, a small contig that has 2500X coverage aligns to another pseudomonas putida submitted by this same lab in Oct 2019.
It also has 100% alignment to Pseudomonas plasmid reported by the same lab.
This actually makes sense. The plasmids are at 2500X coverage when the genome is at 40X. So when plasmids do exist in these very Pseudomonas strains in question their coverage comes back at 2500X, not 2-4X.
One hypothesis that favors these plasmids being from the date of collection rests on Gentamicin providing some selection. Many P.aeruginosa infected patients ‘might be’ put on this antibiotic and provide the required selection for the plasmid to propagate. Someone suggested Gentamicin resistance is missing from the genome. These can be complicated searches as there are many routes to resistance so it is best to rely on industrial tools designed to flag these in genomes. OneCodex has tools for this.

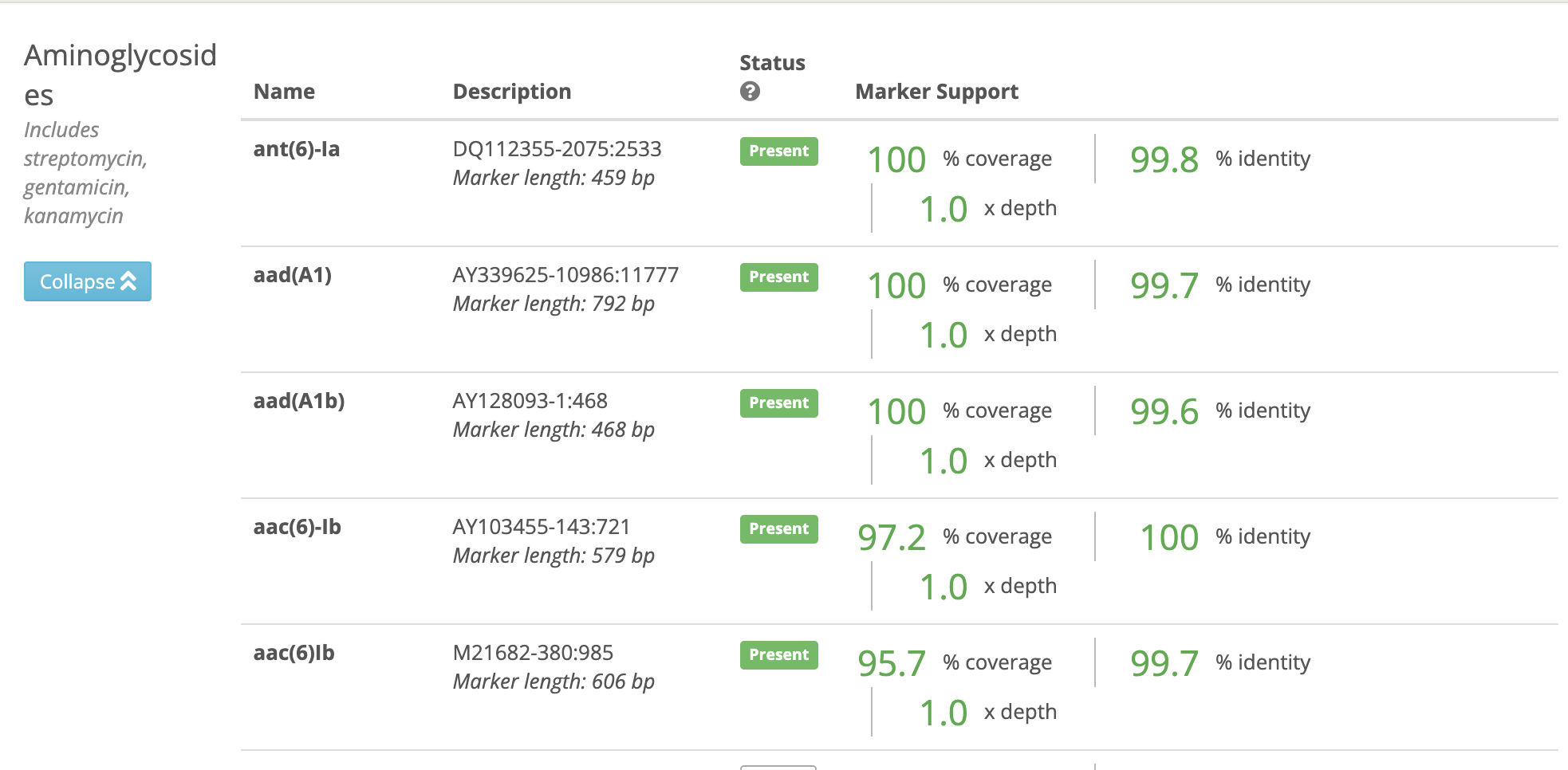
Simply take the P.aeruginosa genomes and remove the contig that codes for the plasmid in question and put the remaining genome through an AMR tool. Note the presence of the AAC(6) gene. So the P.aeruginosa sampled in 2019 is Gentamicin resistant and AmpR resistant and Kanamycin resistant. I find it very difficult to believe that these 4 different plasmids were found in patients surveyed from Urine, Sputum, and BALF. One has a GFP marker in it. This is a research grade plasmid.
In summary, I don’t think the 2019 collection date guarantees these plasmid were present at that time. I think the antibiotics mismatch and the coverage mismatch strongly down weight that hypothesis.
They could have been introduced during sequencing and we don’t know that date.
More developments. I think this thread may have reached a few people at NIH and they have now flagged the sequence as contaminated.
This has of course reinforced the more nefarious hypothesis.

Hard to know since the authors havent provided the date of sequencing.
It very well could be a contamination from a lab that ablated FCS in order to make spike protein from the vector that they could test drugs against.
What a fantastic article!