Compounding Error
Transcriptional and Translational error


This thread on Twitter might not survive long term so I thought I would comment on it in light of a recent paper that deserves it’s own ‘Nerfing The Abstract Award’.
It starts with an important paper that evaluates the Transcriptional error of T7 RNA polymerase used in the mRNA generation for BNT162b2 and mRNA-1723. This paper used the most accurate single molecule DNA sequencers one can use today (PacBio). There is an even higher quality version of this chemistry known as HiFi that would improve the study but this is the best study I can find to date that accurately captures the error expected from incorporating modified nucleotides with error prone RNA polymerases. Specifically this measures the error one sees transcribing an mRNA from a DNA template like the DNA plasmids that encode the mRNA vaccines.
These author see a 1:6,000 error rate (~130-300/Million) and this error rate escalates (1:3000-1:4000 )with the substitution of Pseudouridine or N1-methyl pseudouridine. The vaccines are ~4,200 bases in length so this implies 1 error in every vax molecule and you are injected with 2 shots at minimum which amounts to ~13T-134Trillion of these molecules with vaccination (30ug Single use Pfizer shot vs 300ug (3X Moderna shot). Of course, some people get addicted to these needles and have taken 4 or more shots but I think they represent a very fringe minority of hypochondriacs to extreme virtue signaling politico’s.
To confirm these calculations you can use the NEB RNA calculator.
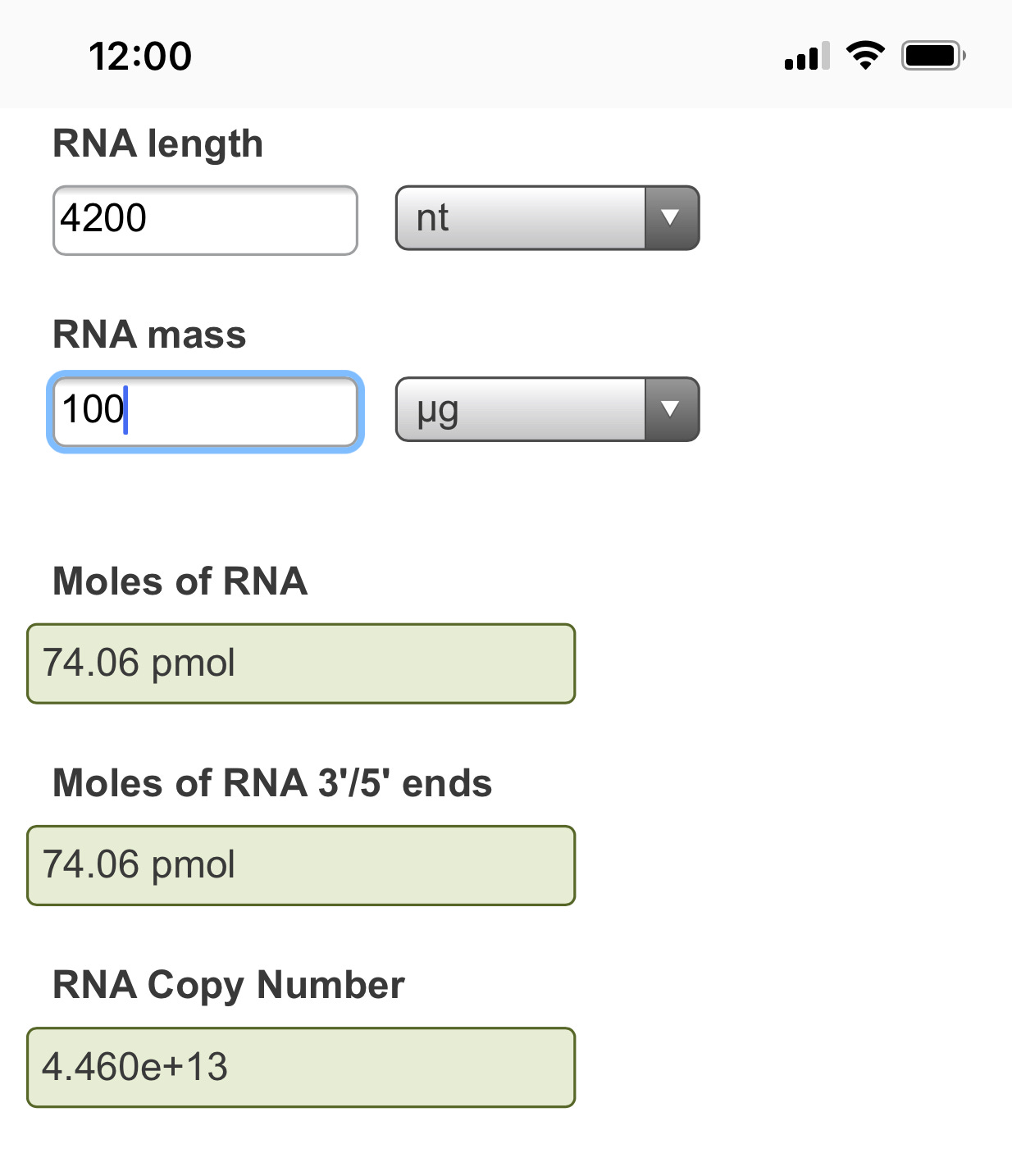
Error rate measurements below are from Chen et al and their team at New England Biolabs.

The polymerase makes errors using this base because it has a different shape than Uracil. There is an extra hydrogen bond that can form with Pseudouridine. N1'methyl pseudo U replaces this hydrogen with a methyl group that changes the bases water solubility and creates stronger base stacking compared to Uracil. This altered base stacking ultimately means the base is a ‘stickier’ base and raises the melting temperature of the double stranded nucleic acid.
This ‘stickiness’ also confuses the ribosomes that must read this RNA and translate it into protein. This is where the transcriptional error compounds with translational error. The N1-Pseudo Us throw sand in the gear of the two main cellular fidelity systems, Transcription and Translation. They likely have influence on miRNA biology and telomere biology given the role natural RNA methylation plays with the PUS class of enzymes.

Much of this detail is archived on a Threadreader page . Twitter will likely memory hole it.
Very recently, A paper emerged that finds similar error rates with a less accurate sequencing methods but the authors define this as being ‘faithful’.


This paper is a Nerf factory and I’m going to cover some of the language that is used while the data supporting those strong terms are buried and obfuscated in the text and figures. The paper has heavily distilled ‘Highlights’ that require data excavation to find their support in the paper.
Let’s start with the paper ‘Highlights’. This is always where the Nerfing is in overdrive as the authors try to make their paper readable and of interest to a general audience.

You’ll notice no quantitative information is put forward for the term ‘accurately’. The graphical abstract depicts ‘faithful protein products’. The summary is equally evasive and vague regarding what the authors mean by these terms.

The phrase ‘does not significantly impact translation fidelity’ is usually reserved to describe a measured P-Value yet none is given. The term ‘marginally’ is undefined. The introduction doesn’t define these terms either.
In attempt to defend these conclusions, the authors spend a fair amount of time on distraction and smoke screen assays to make the paper appear to be supported by multiple methods, all designed to not actually quantitate the problem they claim to address.
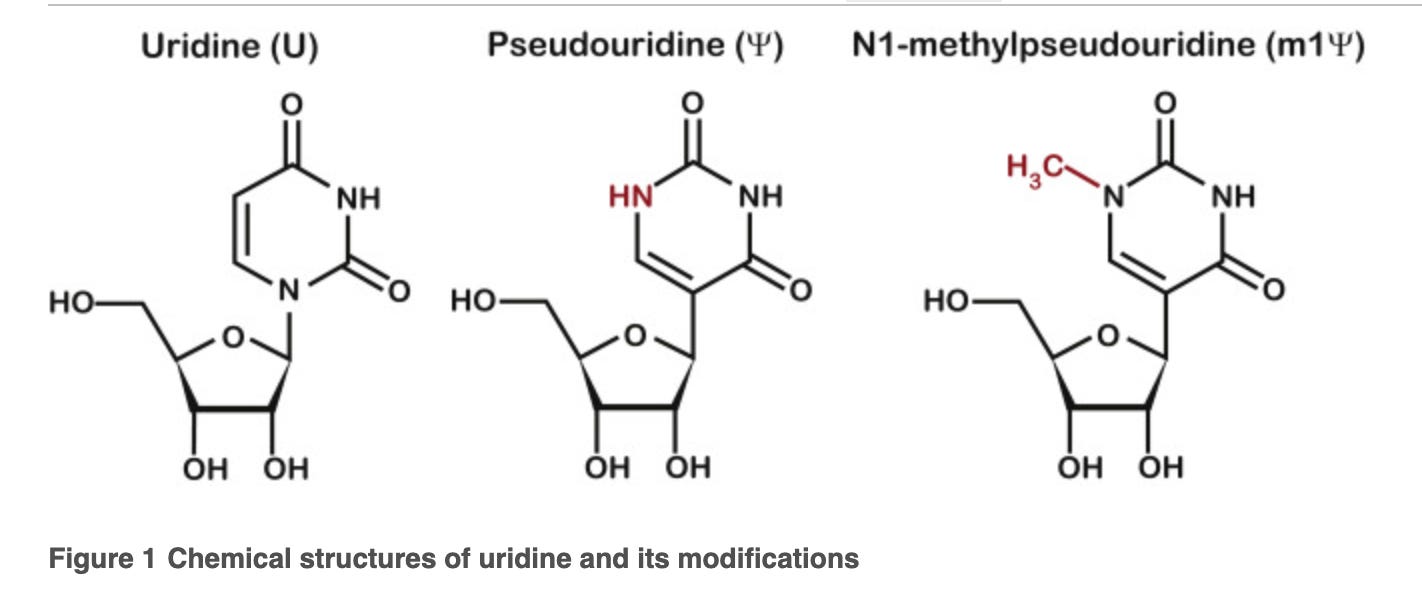
The first smoke screen is to make 3 model templates (not related to the vaccine) with a SINGLE base replacement in a 3 base codon with the above 3 nucleotides. This is a major Nerf. The mRNAs in the vaccines have ALL uracils replaced with N1-methylpseudouridine. If they wanted to steel man their own papers they would ask what happens with the codons that have 2-3 replacements? How about when the entire mRNA has replacements? Polymerases are more error prone when incorporating across secondary structures and this base replacement is known to raise the Tm over 10C with just 4 replacements.
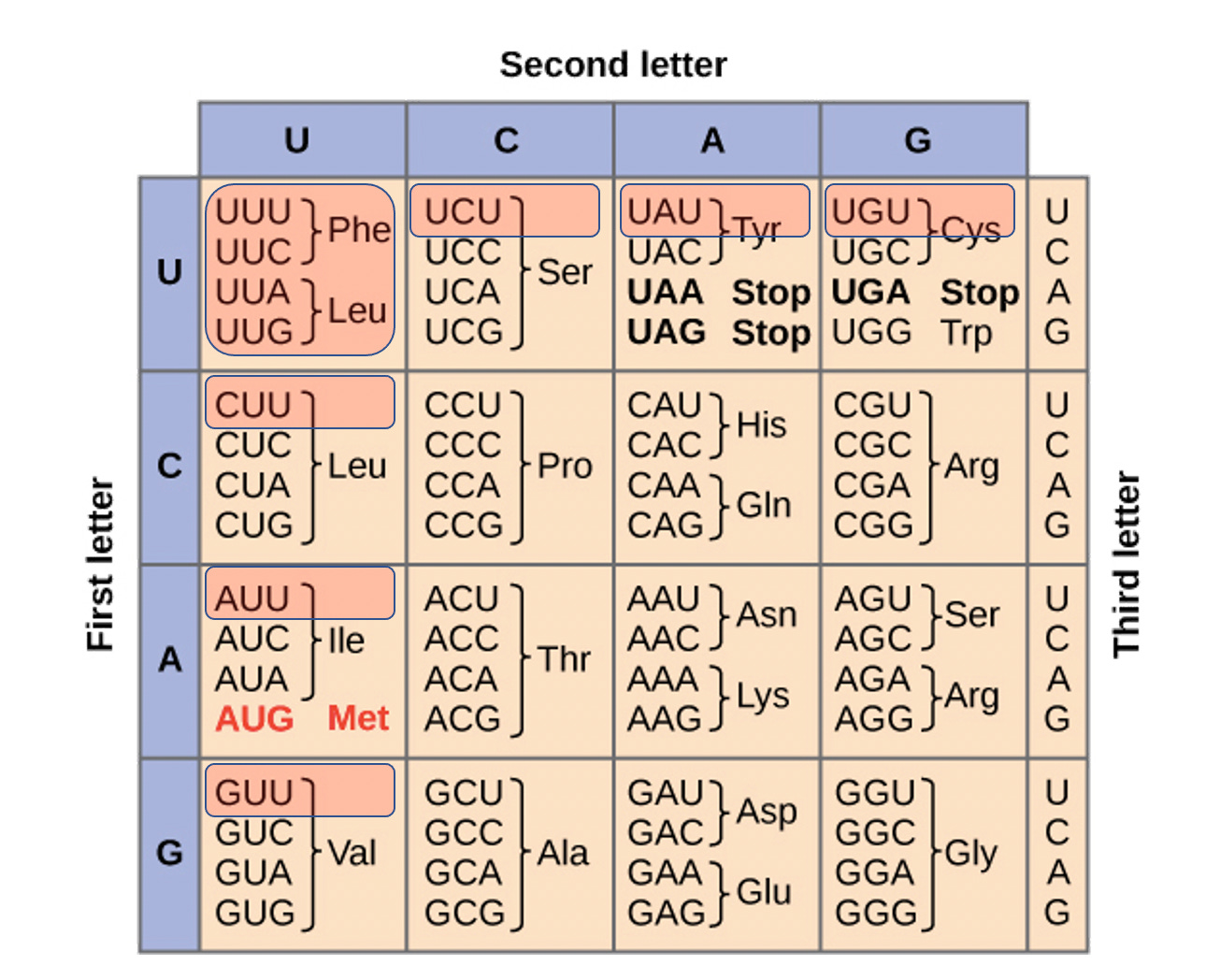
Codon Table Figure- BNT162b2 and mRNA-1273 have global Uracil replacement with N1-methylpseudouridine. The red highlighted codons contain more than one uracil replacement and are most at risk of translation error. This entails Phe, Leu, Ile, Val, Tyr, and Cys.
As you can see, there are many in the genetic code and to simplify this to a single base replacement when the mRNA in the vaccines have all of the uracils replaced with these is a reductio ad absurdum.
To this point, the amino acids most at risk for translational error are spread across multiple classes of amino acids. Leu, Phe, Ile, Tyr and Valine are all Amino acids that code for hydrophobic side chains. Cysteine is an amino acid that can form disulfide bridges and can have a big impact on tertiary structure of the protein. Serine is a polar amino acid.

This is an important detail. When tools like SNPeff are evaluating the impact or severity of a given nucleotide change, a heavy penalty is applied if the SNP induced amino acid change then switches amino acid classes.
So any model that distills “accuracy’ from a Nerfed single base model of the problem is straying into political territory. This is unfortunate, as I really like the work these authors have performed but when I see such biased summarization of the their own data, I lose all trust in their honesty. I went straight to the funding sections and conflicts when I saw this. More on that later.

Let’s look at some of this in detail. Always be suspect of bold claims where the data supporting such claims is relegated to the supplement. They only alter the 1st base in the codon and one can clearly see different kinetics with each base (Squares, circles and triangles reflect each base- highlighted in red on the figure). When ever you want to suppress a difference visually, you can convert the Figure into a log scale on the Y axis (figure K and J- green highlight on scale) and claim there isn’t much difference between these substitutions.
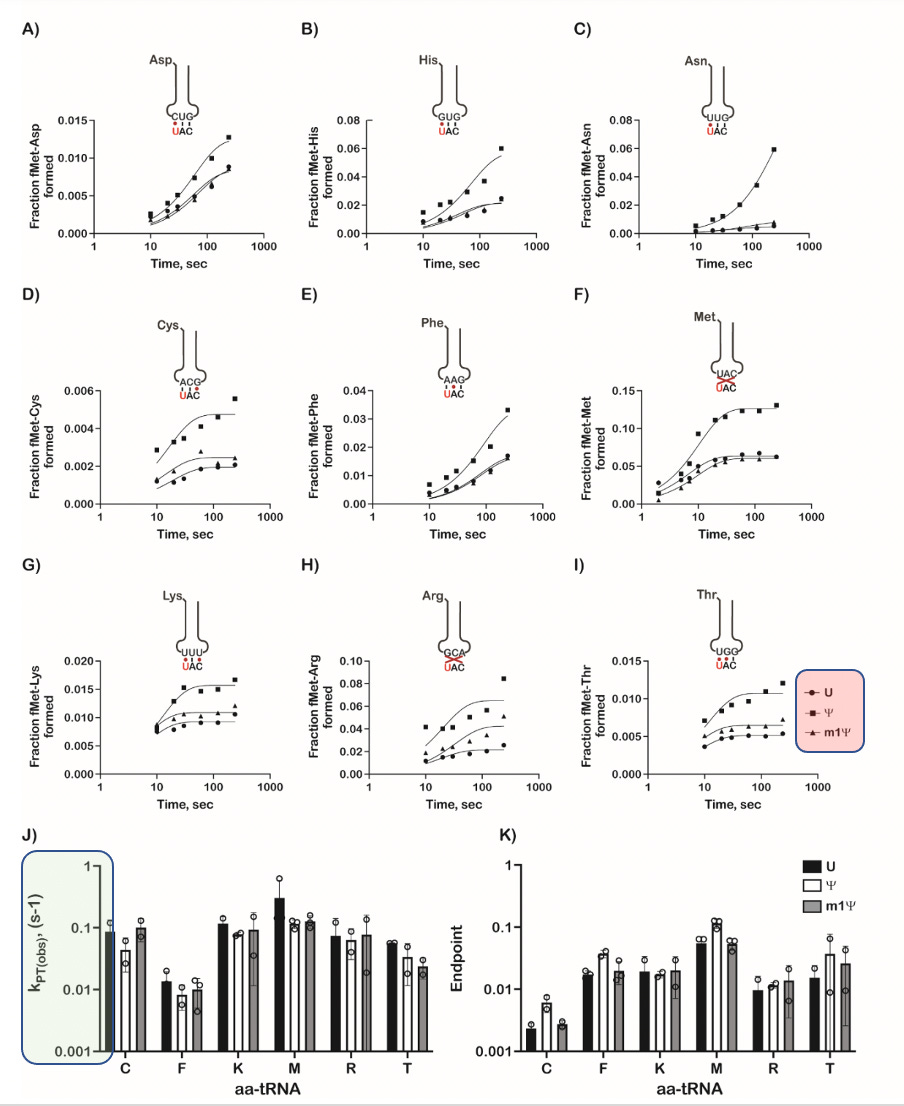
The paper also depicts ‘modest but significant’ reductions in the rate of protein synthesis. This is Nerf language where confidence intervals and p values are preferred. They then change the topic to protein yield to smoke screen this significant finding. Zero mention of the Ribosomal Pausing and the impact this may have on protein folding.
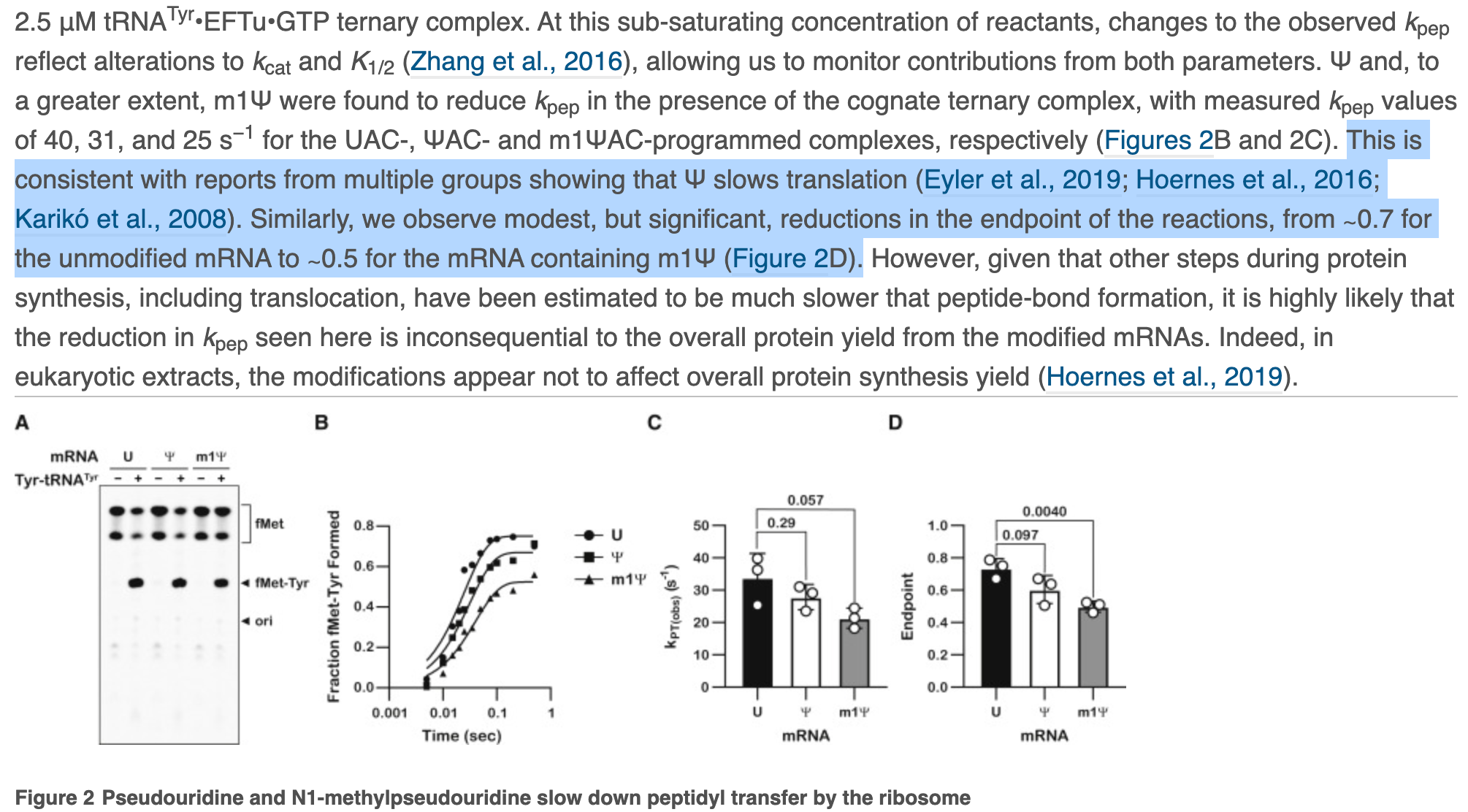
This is a major oversight and whitewashing of results. They should address Postnikova et al. If you alter the rate of translation, you alter protein folding and this is known to effect the structure of Spike protein folding.

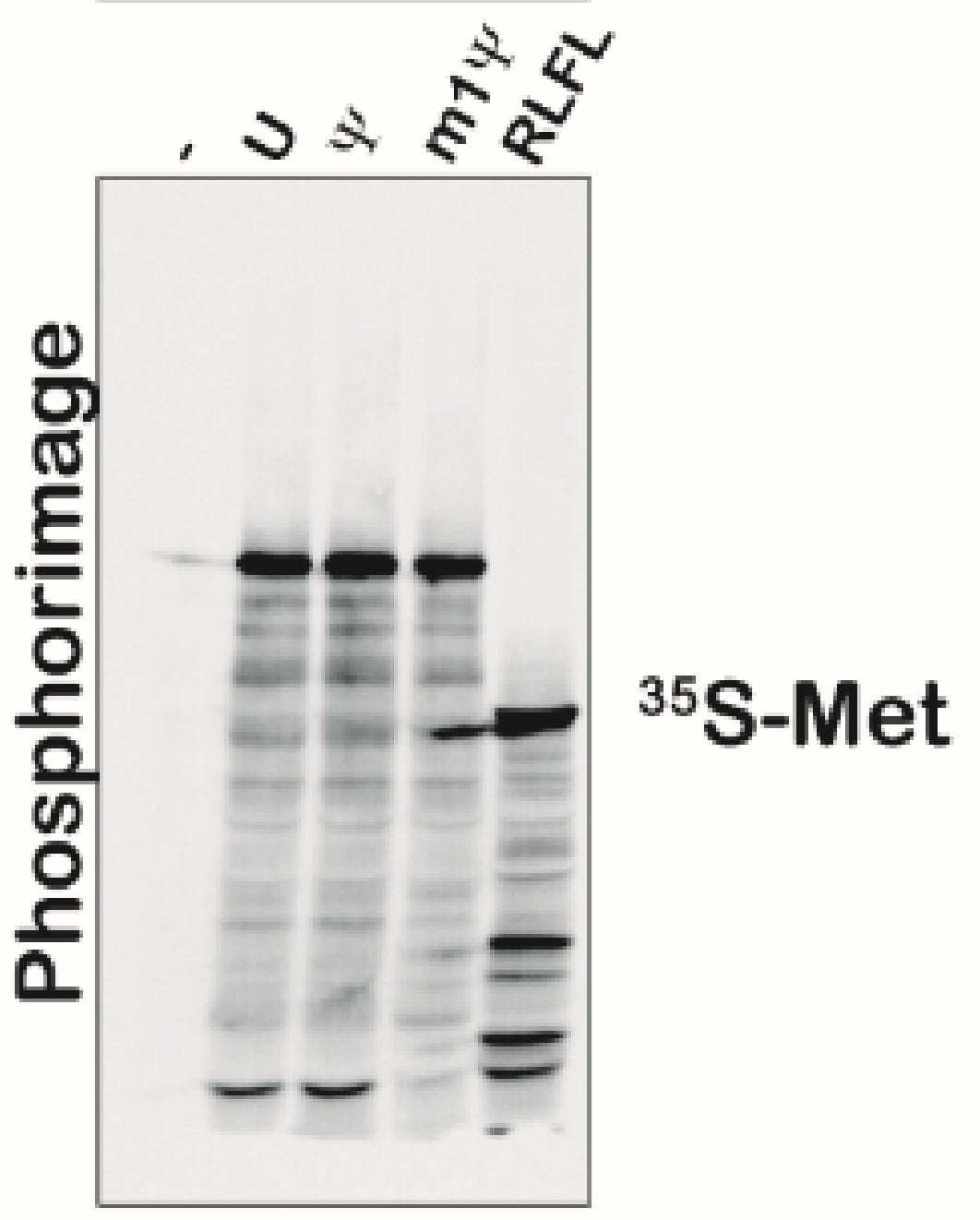
Moving on to Figure S5, we have phosphoimager data with different banding patterns that are discussed as equivalent. Maybe there is some lane to lane bleed but the m1Psi appears to have more background bands.
But most importantly, the claims of accuracy in the Highlights seems to rest on this Mass Spec data buried in the supplement. There are a lot of Non-detects (ND) in the chart which begs the question of what is the Limit of Detection (LOD) of this assay. The Y axis implies 10% translational error at F112Y and some differences at other locations. There are differences amongst the bases.
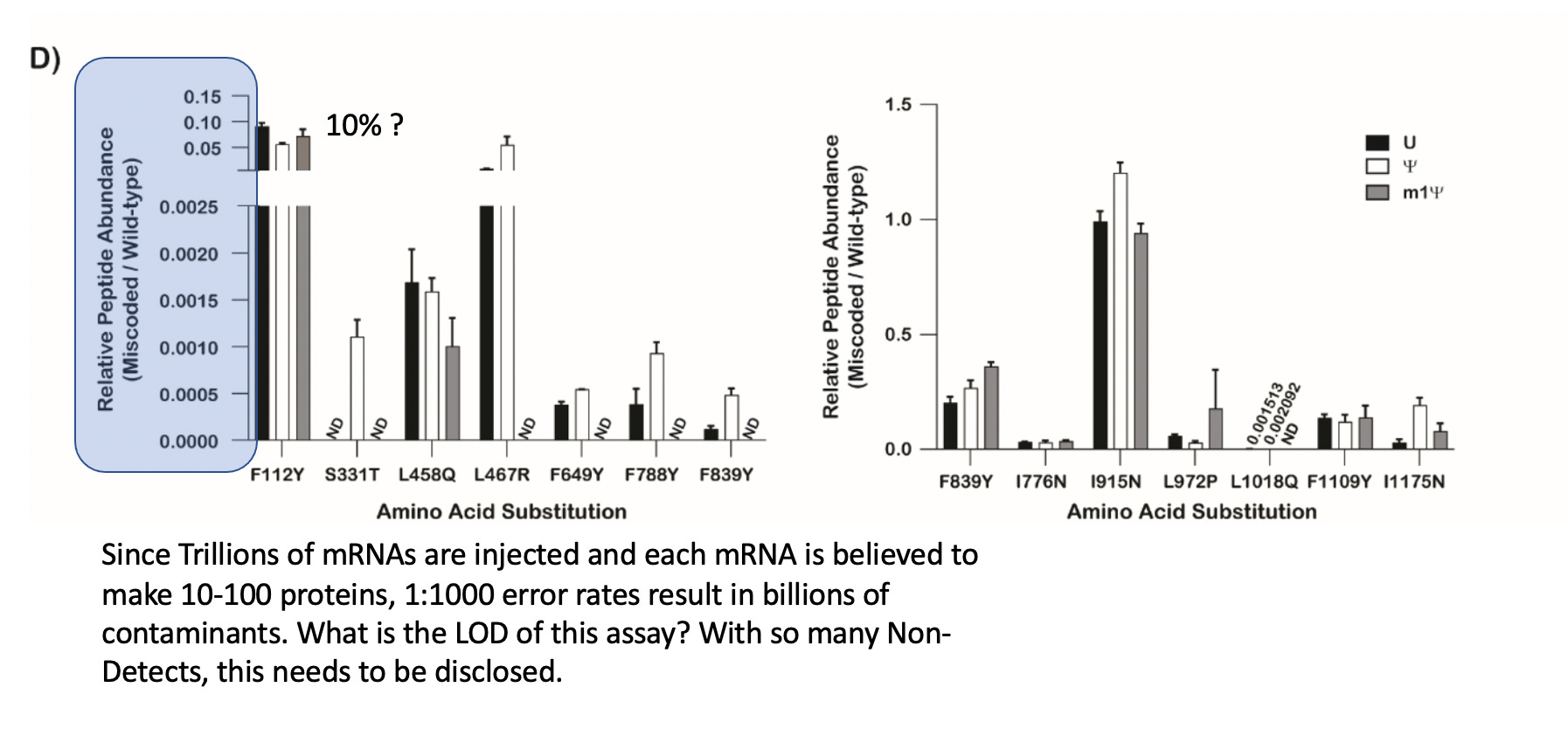
Text describing these data suggest 50% increase in error is ‘modest’.

We have more assurances of accuracy without much quantitative language. This experiment is again only looking at one codon (UAC) with single base replacements at the 1st U in red against 19 amino acid tRNA sequences. What about the other 63 potential codons?
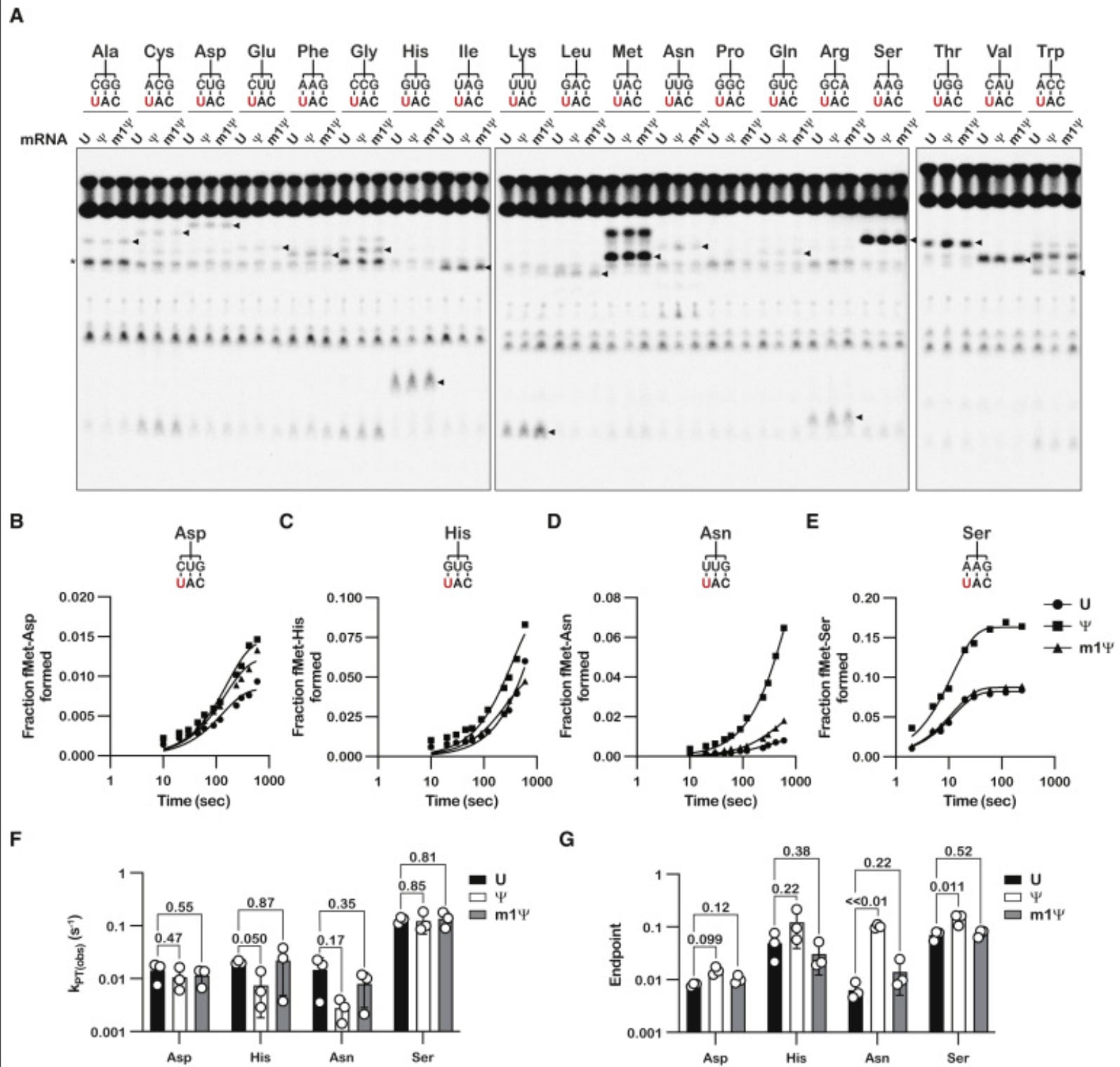
The papers they cite here are good reads. Fernandez et al. we have cited in previous work. When you a get a result that is divergent from this much literature, you might want to use less definitive language in your Highlights section.
It is a bit like “Despite all of these other papers finding accuracy problems with these bases, when we squint at this problem with our irrelevant model, we see no sins”.

The manuscript puts much of the quantitative data into Supplemental Tables. The links provided don’t go to these tables, so I’m being a bit short given the language used and lack of tables one can comb through to decode their rose colored language.
This type of language looks like subterfuge. If you don’t offer up a LOD for your assay and you have 39% coverage of the right peptides in your study, I’m going assume you have no depth of coverage to find 1% errors and you thus don’t care about them. Yet 1% translational error with trillions of expressed proteins, can amount to billions of proteins with error. Keep in mind, this sequence has a super-antigen in it so we want to avoid mutagenesis reactions on super antigens as making them worse would be a real bummer. How much should we tolerate, some ask? I think the tolerance should be near zero given they are being injected by force, without liability and now into children who don’t actually clinically benefit from them. If these were voluntary shots, knock yourself out. To each their own but let’s be transparent about what it is.

Lets see if the folks know how to DNA sequence.
First fowl- Why on Earth are they studying Reverse Transcriptase error rates when the vax manufacturing process doesn’t entail this? The process utilizes a DNA template from a plasmid and a RNA polymerase. I’m confused by this whole section.
And here comes a big fat Nerf Fast Ball. A Lemon of the study turned in to pure lemonade. To summarize, since the real world has 100% uracil replacements, we made a model that would ensure our pre-ordained conclusions deliver Safe and Effective language.

They then waste the readers time with an irrelevant primer extension assay and then admit it is irrelevant since there isn’t competition in the assay with all four nucleotides. Somehow this non sequitur is supposed to help you believe these claims. If you can’t fool someone with 1 straw man, erect 5 others.

We are back to model templates that don’t reflect the vaccine here.

And a major swing and a miss. To accurately segregate library construction errors from your polymerase errors, the sequencing industry has adopted Unique Molecular Identifiers or UMIs. These are known DNA barcodes you ligate to your target sequence and they help to itemize where in the process the errors are being introduced. This is often required in library construction steps that require DNA polymerases to convert the RNA into DNA for DNA sequencers to read. We do not have accurate RNA sequencers. Oxford Nanopore is on the bleeding edge of this field but their current accuracies can’t address 1% heteroplasmy questions. As a result most methods convert the RNA into DNA with RT-PCR and this process can introduce its own polymerase errors. But by gluing a unique DNA barcode onto every molecule before any replication is performed, you can better itemize the source of the errors.
To their credit, they tried to use UMIs. I don’t understand why this failed. I’ve used these and I can only imagine they failed because their templates were too short. It appears these are not mRNA vaccine molecules. I downloaded their reads from the SRA and mapped them to C19, Pfizer & Moderna vaccine sequences and failed to get anything to map with bwa mem? I suspect they are model templates again? They combined this failed experiment with their last primer-extension failed experiment and then distilled ‘accuracy’ in their ‘Highlights’ section? Given the PacBio work that precedes this (Chen et al.) ,which I fail to see cited anywhere in this work, I’m a bit perplexed.
Do they work for Pfizer?
It is not that overt. But in a world of Faucism, one does have to look carefully at how tightly PIs in the funding chain of the NIH beg for more money. I can find NIH grants for the PI. His CV suggests he is waiting on potentially more funding from NIH and I bet he has forwarded this recent paper to the study section/Council Review to seal that deal. Good cookies will be handed down from on high.


As many of my readers know, NIH hold patents on these mRNA vaccines and they have been less than honest with the public describing how natural they are. Natural mRNAs are not patentable, so one side of the NIH office has convinced the USPTO that they are inventive and non-natural while the public facing side of NIH is preaching the exact opposite tale. They are going to cash shower any work that underscores safety of these mRNAs and I’m willing to bet this Nerf awarded paper will get its next ROI grant.
Their paper constitutes a lot of work and its very needed in this day and age. I do not come to the same conclusions as they do as we fundamentally have a different opinion of acceptable slop. The paper does very poor job describing their limits of detection for each assay so it is unclear if they can even see 1% error rates. In the sequencing world, 1% error was considered dirty. Most of these DNA polymerases have 1:1,000- 1:100,000 error rates for discrete sections of the DNA sequence (Q30-Q50). We have Log quality scores to describe the likelihood of error at each base. This paper leads with accuracy but offer little quantitative defense of the term in its splashy wake.
Unfortunately, they may have to play the game to get funded and the readership just needs to get wise to the bias this introduces to public science communication. Never take the Abstract and Conclusions on face value. Always look to see if the authors bent the interpretation of the data to appease the funding source.
Over the course of the last couple of years the way I read scientific papers has changed. I used to read the abstract, get juiced up, and dive into the details of the paper. I now read the abstract, cognisant of bias, then immediately look at the author bios and funding sources! Very good summary Kevin and the fact they don’t cite Chen et al is a genuine smoking gun. It’d be like coming up with a new theory on gravity but comprehensively failing to cite any work by Einstein!!
Most of the paper way above my level of technical understanding, but you still managed to make a complicated subject not only logical, and interesting, but also light, exciting and, dare I say it, entertaining. A joy to read. Thank you.
At this stage of the Pandemic Saga I’m drawn more and more to this kind of granular analysis of the nuts and bolts, rather than sweeping generalised opinion pieces. Ultimately you have to follow the evidence trail.