

DNA fragments detected in monovalent and bivalent Pfizer/BioNTech and Moderna modRNA COVID-19 vaccines from Ontario, Canada: Exploratory dose response relationship with serious adverse events


Our most recent PrePrint on the topic.
TL/DR
All vials exceed the guidelines by orders of magnitude using Fluorometry. All vials are under the guidelines using qPCR for single dose. Over once you take many. XBB1.5 still has the problem.
Multiple tools used to triangulate on a quantitation: Fluorometry, qPCR and Oxford Nanopore.
How does Fluorometry work?
It uses a intercalating dye like SYBR Green. This is a DNA specific dye that binds to dsDNA and becomes fluorescent only when bound up in the DNA minor groove. It has some cross talk with RNA which we will touch on later but you will notice it doesn’t have a size limit like qPCR

How does qPCR work?
qPCR requires a >100bp amplicon and thus cant quantitate DNA under this size range. The amplicon requires a 25bp Forward Primer, 25bp Reverse primer and a 25bp probe so its difficult to get amplicons under 75bp. We used 105 and 114bp amplicons to measure ori and spike in the vaccine.

Pfizer uses Fluorometry and UV Spec to measure and inflate the RNA. qPCR deflates the DNA value as it cant measure the DNA below the amplicon size.
Why does this matter?
Lets look at the size distributions of the DNA in the vaccines. Using Oxford Nanopore we can get a glimpse of the sizes. This is better than any electrophoresis system as we get the single molecule sequence information for each molecule present. Not just some glow on a gel but the actual sequence identity of every molecule that makes up the sample.

There are some limitations to this system related to how you purify the DNA before ligating the Oxford Nanopore (ONT) DNA sequencing adaptors to the DNA you are about to sequence. The ONT system needs a piece of DNA that is tethered to a motor protein that feeds this DNA at a ATP controlled rate into the pore. 1st Step of ONT sequencing is to glue this piece of DNA with a motor protein onto the DNA you want to sequence with a ligase. This ligation step can bias the size distributions present.
The largest source of this bias is from a technology I know well. Its a commonly used DNA purification tool known as Ampure. I developed this tech at MIT and it was the foundational technology of Agencourt Biosciences. It was used extensively in the human genome project and has one really valuable feature to it.
You can control the size distributions of the fragments it captures to magnetic beads just by changing the amount of reagent you add to your sample. No more columns with defined size cutoffs. An open source size selection technique that affords easy open source adaptation to various application in genomics. Beckman Coulter came to acquire the company as this tech took off and now is found throughout all Next Gen sequencing applications.
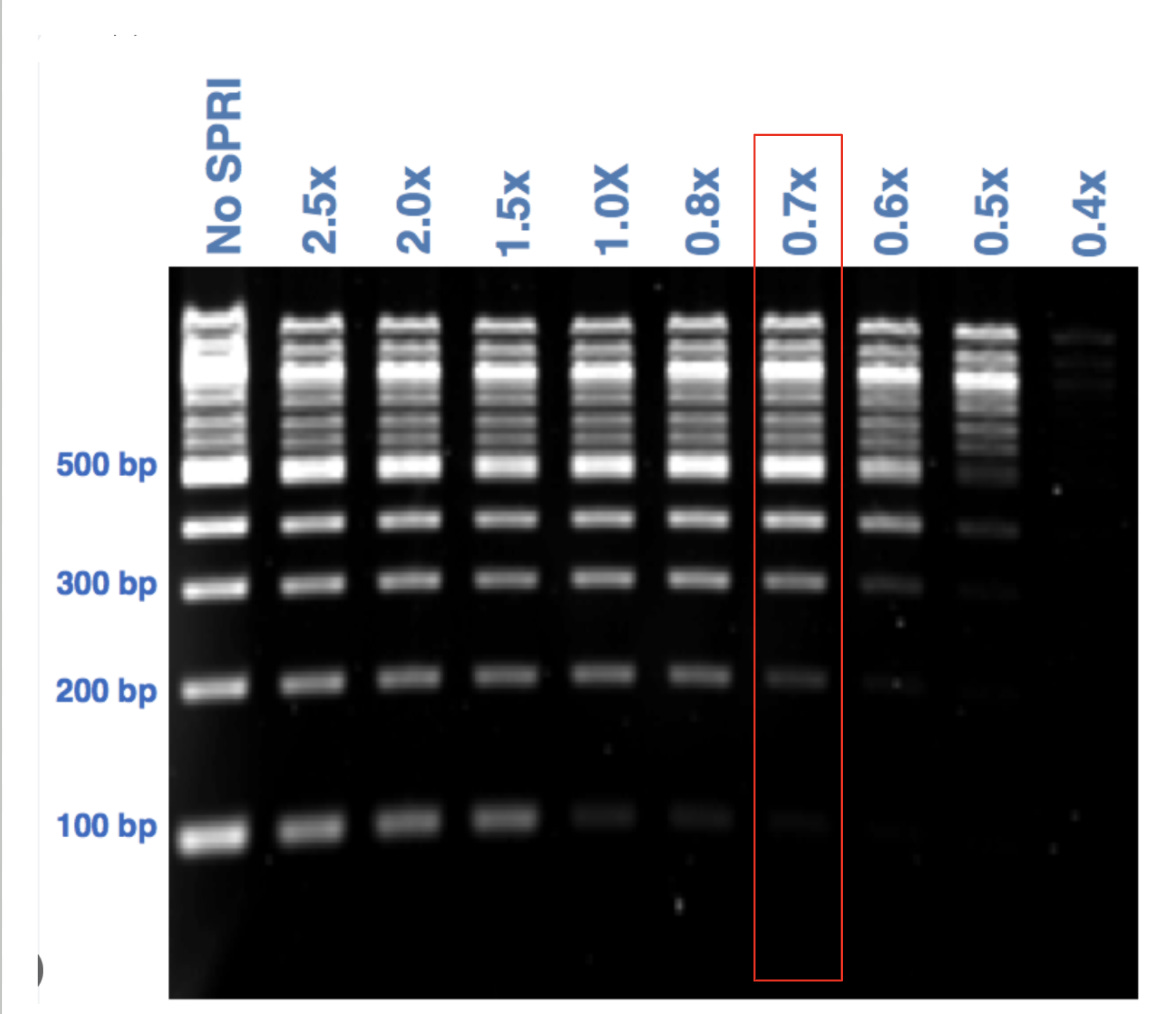
Since most people use ONT to get long reads they have a default protocol that uses 0.7X Ampure to get rid of the DNA shrapnel in DNA sequencing libraries. This cuts out the fragments 150bp and smaller. So our Read Length histogram above has to be considered in light of this front end filter. Even with this extreme cut off, we still see reads under 100bp which means the original small material has to be at very high concentrations for it to sneak through this.
Dr. Phillip Buckhaults expected this and used 2.5X Ampure before his ONT run and his mean read length is lower than our 214bp mean.
The DNA denialists like to point out that the FDA doesn’t consider DNA under 200bp to be a concern. That’s not entirely true. Kilnman et al is a good read for FDA guidance.

You will also notice kurtosis in the read length distribution. A long tail in the read lengths with some reads at 3.5Kb (See blue line in the below plasmid map) in just 865 reads. This is a very shallow sequencing survey. Deeper sequencing will undoubtedly find 7kb reads.
The next thing we tested was if this DNA was wrapped in an LNP. If you followed this substack, this is old news to you.
With the DNA measured two different ways we observe log scale different answers, likely due to some qPCR and ONT blind spots for small DNA and likely due to Fluorometry having some cross talk between DNA and RNA. Some RNase experiments are underway to sharpen this but we don’t really need to nail it. The two methods correlate and we have adverse events that track with these quantities.
And thats exactly what
did. She charted the AE against the DNA mass.
Charted another way to normalize for a proxy of the number of doses given… Dr. Wiseman came up with this.

This is exploratory. This is what the detractors of VAERs always claim its meant for. Its not proof of causation but it is hypothesis generating.
So we don’t have a lot of datapoints. But we do have knowledge of other peoples data points. Dr. Buckhaults lots are high in DNA and high in AEs. FL8095 used in our study from April is also high in DNA and high in AEs when charted on this graph. FL0007 we also have some data on that is high in DNA and high in AE. More on this later.
A little housekeeping for those who have criticized our qPCR assay.

Factcheckers got scammed by a basement dwelling debunker who claimed our PCR efficiency failed. 94-99% efficiency is within MIQE guidelines. Debunker debunked and the lab work to do this took less time to generate than his 30 minute video. This was performed by Diamond Hands Dr. Speicher
. Not my hands. Independent validation.Another thing we learned from Dr. Buckhaults… He noticed a non-linear response to LNPs directly spiked into qPCR. What does this mean.
PCR jocks like to run serial dilutions like college kids love serial parties. If your PCR doesn’t show a 3.3CT offset between Neat vs 1:10 vs 1:100 vs 1:1000 vs 1:10K etc dilutions then you have a PCR inhibitor. This can in fact be seen with Neat spike ins of the vaccine.
It is subtle but its enough of an affect that it skews your quant of very concentrated vaccines. Some vaccines come as 5X stocks that require dilutions before injections and there is a litany of VAERs reports from Pharmacists that forgot to do this. This is another confounder in our data. Lots that require dilutions have a higher tendency for adverse events as there is an additional step the pharmacists can forget and easily give a child 5X the dose (purple cap Pfizer).

The net result is that you should use 1:10 dilutions to ensure you are not experiencing PCR inhibition with high concentrations of LNPs.
Why are we torturing you over Fluorometers vs qPCR? Because this technological discordance is a mechanism for regulatory arbitrage. Use the best tool that inflates the RNA and the best tool to deflate the DNA number and you can pass any regulation you want. They wouldnt do that???
Yup.. RiboGreen is a Fluorometric assay for the RNA and they use the more conservative tool (qPCR) to the measure the DNA. This should really be done with the same technology for both nucleic acids. They already had the qPCR assay made to measure this and simply needed to use an RT polymerase to get the RNA+DNA value. qPCR delivers the DNA value. With those two measurements they would have normalized data that uses the same tech for both measurements. Regulators didn’t see this bean under the coconut game.

The other critique thrown at us is the ‘Elf on the Shelf’ hypothesis. Some how, if these vaccines escape the holy hands of the manufacturer an Elf can put the exact plasmid disclosed to the EMA in vials from Canada , Europe, Japan and the US. They can do this without disturbing the tamper evident seals on the vials.
It’s an amazing technology that borders on 5G nanobot sci-fi material.

These vaccines have a documented provenance and cold chain.
But this was all a smoke grenade to spread FUD which was already addressed in a previous SubStack. We have tools that can measure the RNA integrity and decay of old vaccines. Its a non-issue.
The net result…

I think the most hilarious aspect of this project is watching the factcheckers choke month after month while scientists hit the bench and actually advance the story. Its worth collecting these vaxDNA debunk obituaries.
They started by claiming it didn’t leave the arm.
They followed up with it doesn’t last more than 48 hours (plasma, breast milk and heart tissue say otherwise)
They doubled down claiming there was no DNA.
When replicated they claimed it didn’t enter the cell
When shown its in the LNPs they claimed it didn’t reach the nucleus
When shown the SV40 enhancer moves DNA into the nucleus in hours they claim it doesn’t matter.
Where will their artful back peddling lead them next?
Its hard to know given the neurological complications these vaccines can induce.

Thank you for all your work
question is why use plasmid with SV40 elements as template? Its been many years, but we used similar plasmids for stable transfection of mammalian cells. I am thinking they found higher transcript production off this plasmid than the typical runoff plasmid (maybe they screened a bunch of in-house clones). We found transcript levels can vary depending on the background vector Or maybe someone screwed up and they batched the wrong clone and no one noticed? I would have to believe this is no longer in the latest version?