

Fluorometer and UV spectra of purified Pfizer and Moderna vaccines

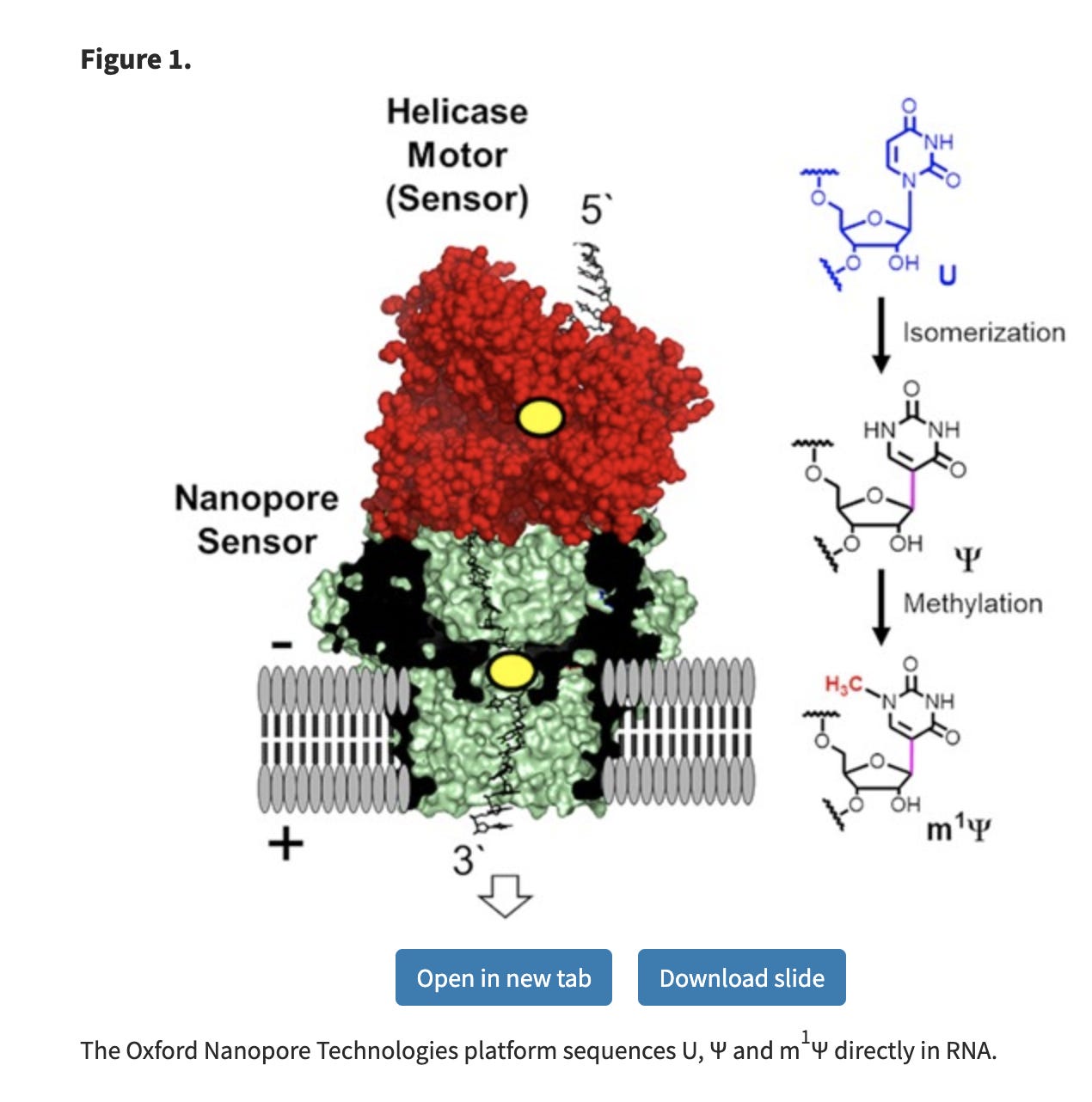
Assessing DNA and RNA concentrations of modified RNAs should be done with every method possible. We are in new territory with these modRNAs as the modified nucleotide are known to creates errors in many sequencers. A good reflection of this is seen in Nanopore sequencing data.
Since the m1Ψ base is chemically different than uracil, it creates a massive elevation in error rates on the Nanopore sequencers. The nanopores sense this as a different base, just like your polymerases and ribosomes, albeit the nanopore error is more extreme.
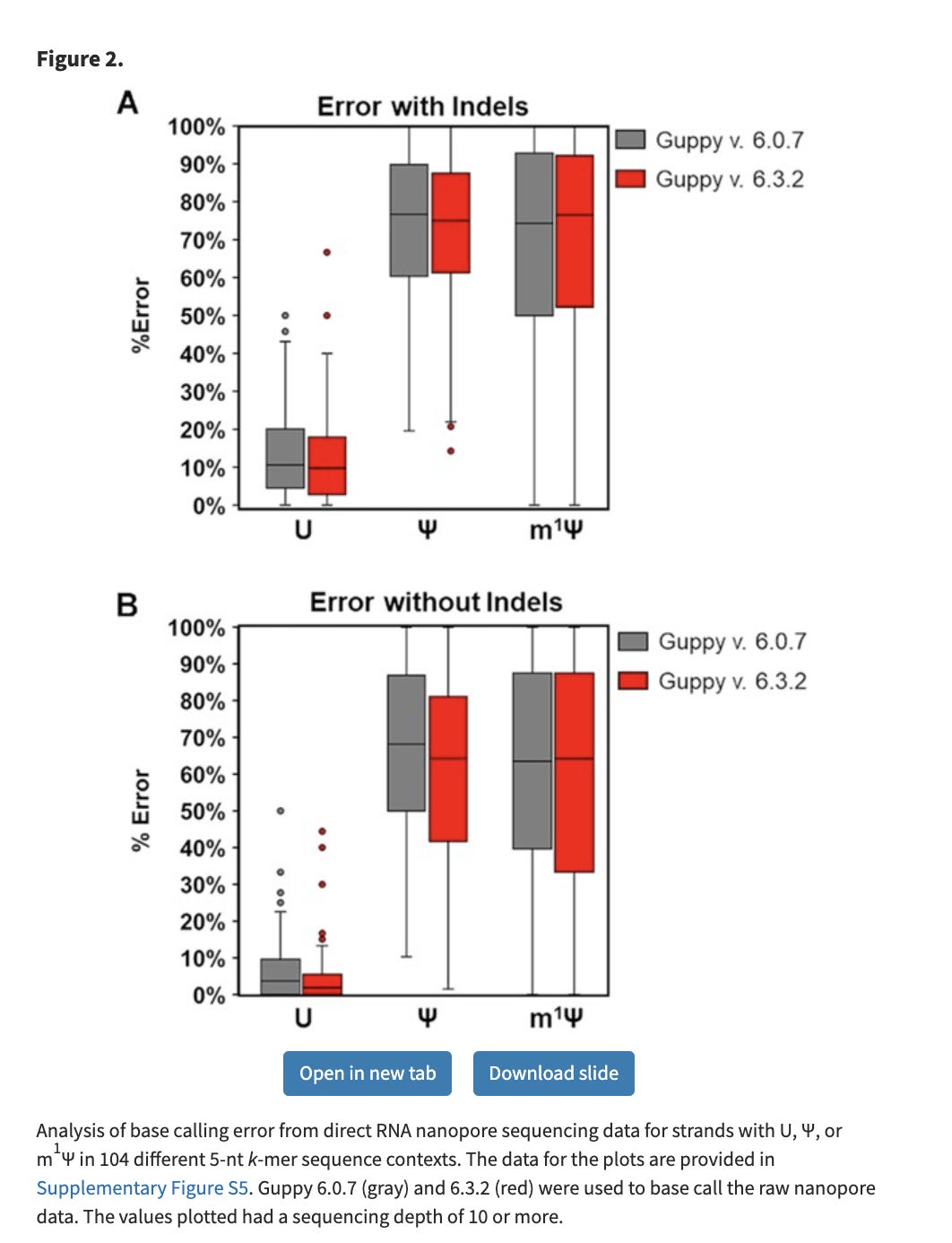
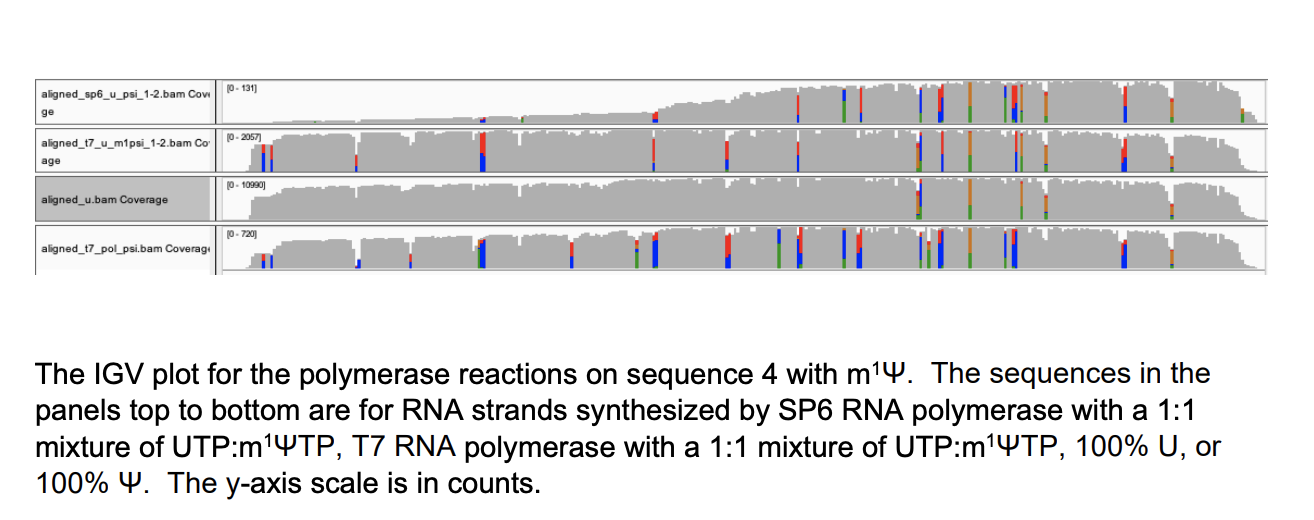
These manifest in False positive SNPs in IGV across the template where ever a U→m1Ψ exists.
As a result of the known artifacts in many genomics tools with m1Ψ, it is helpful to get as many orthogonal measurements on this question to triangulate to a bayesian answer. AnadaAmigo Nate Weymouth suggested UV Spec. This measures UV absorbance of nucleic acids. Residual nucleotides and short fragments still contribute to the signal. But it’s easy enough to run for those aficionados.
Below we have Qubit Fluorometer reading and a Nanodrop UV spectra. The Qubit uses SYBR green I to assess DNA. It clocks in at 1-2.8ng/ul. Thermo’s website has relevant data on the dsDNA vs ssDNA cross talk with these dyes. Its below 10% of the signal.
The UV Spec shows an order of magnitude higher at 13-35ng/ul but it really doesn’t itemize RNA from DNA and is a whole nucleic acid measurement.
The Agilent Tape Station falls between these two measurements at 8-11ng/ul of DNA.

We have multiple technologies triangulating on 1-11 ng/ul and qPCR data before and after DNase and RNase which confirms the DNA and RNA being of the same orderof magnitude.
The next question is how contiguous is this DNA/RNA. Fluorometers and UV Spec will provide signal on 100bp fragments or 7.7Kb fragments. They cannot inform on the contiguity or length of the nucleic acids like the Agilent gel electrophoresis.
AnadaAmigo at-P_J_Buckhaults on twitter suggested I attempt an Oxford Nanopore Rapid Barcoding kit to understand if any of the fragments were longer than shrapnel. These pores do not sequence RNA and we RNase R’d the sample to reduce the amount of RNA in the sample. Unfortunately, all of my flongles were expired but instead of tossing them out, I sacrificed them to the greater good. A mere 10 pores to start and it still managed to barf out a few reads. This needs to be repeated on new chips but its fun to see sequencers spit out 1478 base single molecule reads that are 97% identical to the DNA based vector. These were K114 chips and this is the expected accuracy of the sequencer.
@36780053-928c-4857-8e8e-a50b1271fcd0runid=f99a2335fdfba996a8a26817a22e586b4acb152b sampleid=test read=80515 ch=122 start_time=2023-03-14T19:27:37Z model_version_id=dna_r10.4.1_e8.2_sup@v3.5.1
ATTTTTATTGCTAACTCGTTCAGTTTAAAAAACAATATCACATCTTATATACCATTACCAGGGTTTTCGCATTTATCGTGAAACGCTTTCGCGTTTTTCGTCCGCGCTTCACTTCAACTGCTACTTCCCACTGCAGTCCTACGGCTTTCAGCCCACAAATGGCGTGGGCTATCAGCCCTACAGAGTGGTGGTGCTGACTTCGAACTGCTGCATGCCCTGCCACAGTGTGCGGCCCTAGAAAAGCACCAATCTCGTGAAGAACAAATGCGTGAACTTCAACTTCAACGGCCTGACCGGCACCGGCGTGCTGACAGAGAGCGACAGAAGTTCCTGCCATTCCAGCAGTTTGGCCGGGATATCGCCGATACCACAGACGCCGTTAGAGATCCCCAGACACTGGAAATCCTGGACATCACCCCTTGCAGCTTCGGCGGAGTGTCTGTGATCACCCCTGGCACCAACACCAGCAATCAGGTGGCAGTGCTGTACCAGGACGTGAACTGTACCGAAGTGCCCGTGGCCATTCACGCCGATCAGCTGACACCTACATGGCGGGTGTACTCCACCGGCAGCAATGTGTTTCAGACCAGAGCCGGCTGTCTGATCGGAGCCGAGCACATGAACAATAGCTACAGTTGCAACATGCCATCGGCGCTGGCATCTGTGCCAGCTACCAGGACAGCAGACAACAGGCCCCAGCAGAGCCGGATCTGTGGCCAGCCAAGACATCATTGCCTACACAATGTCTCTGGGCGCCGAGAACAGCGTGGCCTACTCCAACAACTCTATCGCTATCCCCACCAACTTCACCATCAGCGTGACCACAGAGATCCTCCCTGTGTCCATGACCAAGACCAGCGTGGACTGCACCATGTACATCTGCGGCGATTCCACCGAGTGCTCCAACCTGCTGCTGTCATACAGCAGCTTCTGCACCCAGCTCCAATAGAGCCCTGACAGGGATGACCGTGGAACAGGACAAGAACACCCAAGAGGTGTTCGCCCAAGTGAAGCAGATCTACAAGACCCCTCCTATCAAGGACTTCGGCGGCTTCAATTTCAGCCAGATTCTGCCCCAATCCTCGCAAGCCCAGCAAGCGGAGCTTCATCGAGGACCTGCTGTTCAACAAAGTGACACTGGCCGACGCCGGCTTCATCAAGCAGTATGGCGATTGTCTGGGCGACATTGCCGCCAGGGATCTGATTTGCGCCCAGAAGTTTAACGGACTGACAGTGCTGCCTCCTCTGCTGACCGATGAGATGATCGCCCAGTACACACGTGTCCCTGCTCCACGGCACAATCACAAGCGGCTGGACATTTGGAGCTGGCGCCGCTCTGCCATGCCCTTTGCTATGCAGATGGCCTACCGGTTCAACGGCATCGGAGTGACCCAGAATGTGCTGTACAAAAACCCAGAAGCTGATCGCCAACCAGTTCAACAGCGCCATCGGCAAGATCCAGGACAGCCTGAGCAGCA
Italicized sequence aligned with BLAST’d against NCBI

We’ll need to order more chips to repeat this but its good to see contiguous single molecule sequence longer than the short Illumina reads produced to date.
Conclusions
Multiple methods are triangulating on high concentrations of DNA being in the vaccines. Multiple methods observe orders of magnitude more DNA than the 330ng/mg maximum regulations listed at the EMA. Qubit and Agilent systems measure all dsDNA in solution while qPCR will only measure amplifiable molecules that do not have a DNaseI cut site between the primers. Thus the longer you space your qPCR primers, the most discordance you will see with Qubit and Agilent system measurements.
I wonder if someone can work it backwards. Transfect cells with the vax, express the protein, purify the spike and then do amino acid sequencing and determine what possible codon sequences were required to produce the protein
Sounds like a lot of work to do unfunded though
Are you citing Dr Richard Flemming's Jmol conformation? Just curious.