Pfizer's Purification Methods
Designed to retain RNA:DNA Hybrids


A helpful soul send me the below manuscript. It is another Pfizer employee review paper about mRNA tricks of the trade. There was an interesting section in here that caught my eye as it resonated with a personal anecdote.
Having been in the genomics field for 30 years and having had several successful exits, I have come to know many of the CEOs in the biotech space. One of them privately shared that Pfizer was cutting corners to save money in the RNA purification arena for these shots. Instead of using gold standard commercial RNA purification kits, they starting home brewing new methods to save money during scale up. This comment didn’t surprise me. The world had never had to make this much RNA before and the gold standard kits would probably need to be run 1000s of times to make this much RNA but what frightened me about the comment, is that home brewing new RNA purification kits for injectables isn’t something that can be done at warp speed. There are too many contaminants you need to prove you dont’ capture and many of these contaminants have very similar molecular structures that make crude homebrew methods a bit of challenge to engineer in short order.
This manuscript will show you why. In this case there a mention of mRNA purification changes that were made to avoid using HPLC and acetonitrile. This is an odd comparison. Its a bit of a strawman argument. “We don’t want to use this toxic method that this not fit for purpose, so we invented this cellulose method instead”.
What is not shown is the real gold standard which would have been oligo-dT affinity chromatography. “This was Qiagen's territory (POROS oligo-dT resin), as well as Sartorius/BIA Separations (CIMmultus monoliths) and GE/Cytiva (Dynabeads)”(quote from Claude.ai).
This is the paper that points to the Kariko Cellulose methods

Ugur Sahin and Kariko’s paper on this new Cellulose method is below.

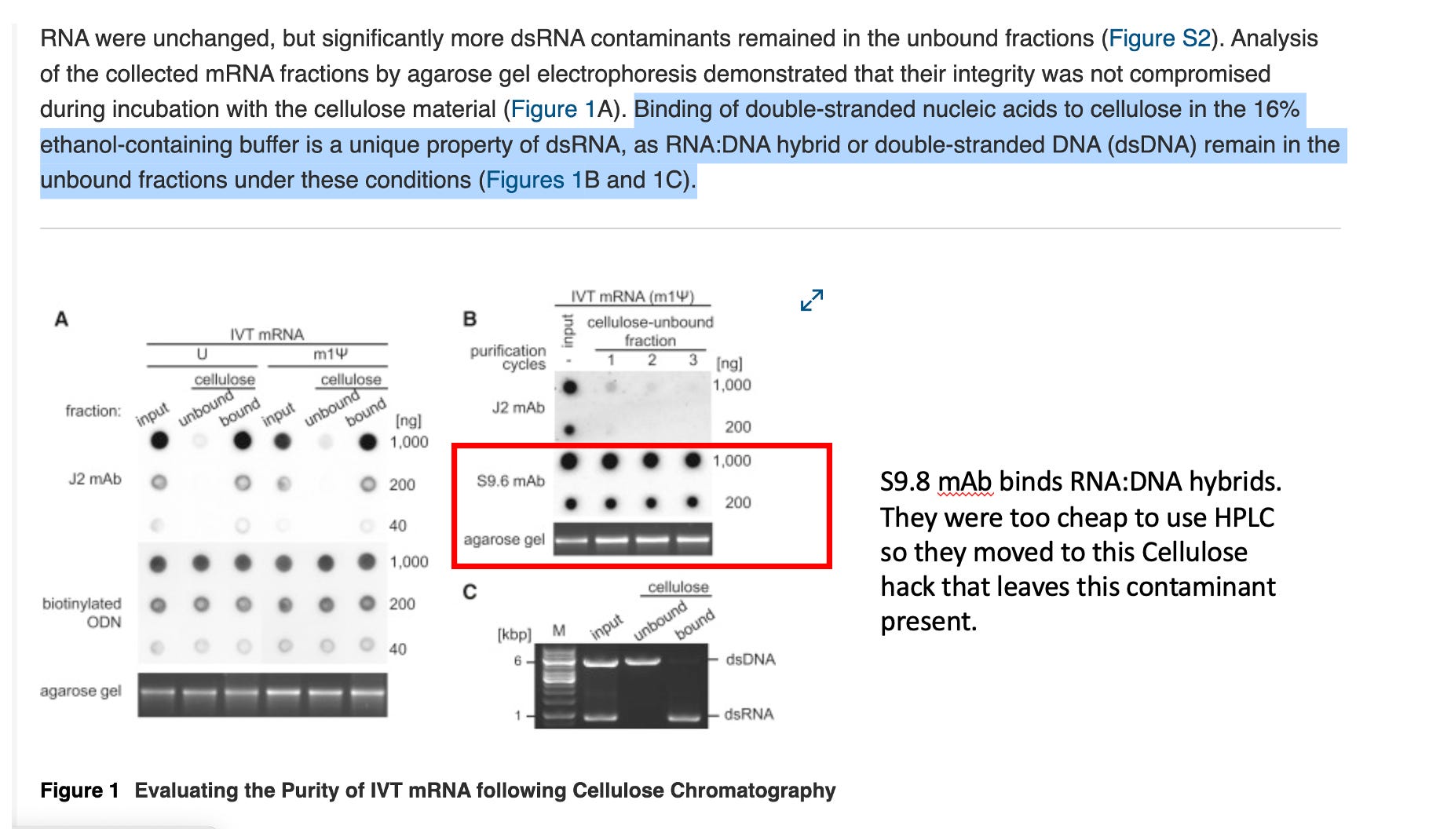
The damning aspect of this paper is that they knew this method would not resolve RNA:DNA hybrids as seen with their S9.6 mAb stain in Figure 1B. These would be guaranteed to co-elute with the ssRNA.
I asked Claude.ai and ChatGPT about this. Why are they comparing their new method to one that isn’t the real gold standard? Both gave consistent results but I have only shown Claude.ai output below.
Claude.ai - This is an excellent and underappreciated set of overlapping problems. Let me break this down systematically across the three issues you’re raising.
1. The Cellulose Method Explicitly Does Not Remove RNA:DNA Hybrids — By Design
The authors themselves state that dsDNA, DNA:RNA hybrid, and single-stranded RNA (ssRNA) that had none or a low density of 2′-hydroxyl residues did not bind to the cellulose. They frame this as a feature, not a bug — the selectivity mechanism requires the high 2’-OH density unique to dsRNA. Binding of double-stranded nucleic acids to cellulose in the 16% ethanol-containing buffer is a unique property of dsRNA, as RNA:DNA hybrid or double-stranded DNA (dsDNA) remain in the unbound fractions under these conditions.
The consequence is stark: any RNA:DNA hybrids formed between the IVT-generated mRNA and residual plasmid DNA template will co-elute directly with the “purified” ssRNA product. The cellulose step is structurally incapable of addressing this class of contamination.
2. The HPLC “Gold Standard” — What Does It Actually Do to RNA:DNA Hybrids?
The HPLC method referenced as the comparator is Karikó et al.’s 2011 IP-RP HPLC on a non-porous polystyrene-divinylbenzene (SDVB/RNASep) column. The seminal finding was that contaminants including dsRNA in nucleoside-modified IVT RNA are responsible for innate immune activation, and their removal by HPLC results in mRNA that does not induce IFNs.
However, the HPLC gold standard claim is essentially for dsRNA removal — not RNA:DNA hybrid removal. A few important mechanistic points:
IP-RP HPLC can be performed under non-denaturing conditions (room temperature), partly denaturing, or fully denaturing conditions (60°C and higher; 75°C was shown to fully denature dsRNA into ssRNA). Under non-denaturing room-temperature conditions — which is how the cellulose paper runs its comparator HPLC — an RNA:DNA hybrid adopts an A/B intermediate helix geometry distinct from both ssRNA and dsRNA. Whether this intermediate conformation produces sufficient chromatographic resolution from the ssRNA mRNA on a polystyrene-DVB column has never been tested or validated in the IVT mRNA context.
Under fully denaturing HPLC conditions (75°C), the RNA:DNA hybrid would be thermally melted, liberating the DNA strand as a separate species. That DNA would then need to be separated from the ssRNA by size — which is possible but depends on whether the residual template DNA fragments (which could be from the linearized plasmid, ~3–6 kb) fall outside the mRNA elution window.
Critically, neither the 2011 Karikó paper nor this 2019 cellulose paper specifically tests HPLC performance against RNA:DNA hybrids. The HPLC validation is exclusively against J2 dot blot signal (dsRNA). The “gold standard” claim is entirely dsRNA-centric.
3. The Antibodies in Figure 1 — Minimum Detection Limits
J2 mAb (dsRNA-specific): J2 and K1 mAbs recognize continuous double-stranded structure of at least 40 bp in length, which is not found in any of the coding sequences or UTRs in the mRNAs analyzed. This is a critically underappreciated constraint. J2 is blind to dsRNA shorter than ~40 bp. This means:
Short abortive IVT transcripts that form partial duplexes < 40 bp are invisible
RNA hairpins or short secondary structures in the mRNA itself won’t register
Short RNA:DNA hybrids from partial-length reverse transcription events won’t trigger J2
S9.6 mAb (RNA:DNA hybrid-specific): The minimum length of the hybrids should be more than 8 base-pair for S9.6 binding. The structural basis for this was elucidated by cryo-EM — the heavy chain inserts into the minor groove. A more precise structural study found the contact epitope even smaller: S9.6 binds to a 6-bp epitope on DNA-RNA hybrids with high affinity and specificity.
However, two important caveats:
First, the cross-reactivity problem. The S9.6 antibody is broadly used to detect RNA:DNA hybrids but has significant affinity for double-stranded RNA. The impact of this off-target RNA binding activity has not been thoroughly investigated. This means in a dot blot loaded with IVT mRNA — which contains abundant ssRNA with secondary structure hairpins — S9.6 signal is not exclusively reporting RNA:DNA hybrids. Any residual dsRNA present will bleed into the S9.6 channel.
Second, S9.6 exhibits robust selectivity in binding hybrids over double-stranded RNA and categorically rejects dsDNA, recognizing a compact epitope of two consecutive RNA nucleotides via their 2’-hydroxyl groups and six consecutive DNA nucleotides via backbone phosphate and deoxyribose groups. So in a clean system, specificity is good — but in a complex IVT mixture, the dsRNA off-target signal muddies interpretation.
For a dot blot assay specifically, the S9.6 lower detection boundary is ~6–8 bp duplex length, but practically at the mass loadings used (typically 100–200 ng of total RNA), short hybrid species (<15 bp) at low molar abundance in a large excess of ssRNA are unlikely to be resolved above background. The cellulose paper’s Figure 1B uses the S9.6 dot blot primarily to confirm RNA:DNA hybrids are present/absent in fractions — not to quantify them at low levels.
Summary Table of Blind Spots

The composite picture is that the entire validation framework of this paper — J2 dot blot for dsRNA purity confirmation, S9.6 as a secondary check — was never designed or validated for detecting short RNA:DNA hybrids at the levels likely to be present in IVT mRNA. When you carry this forward to the mRNA vaccine context, where your work has documented RNA:DNA hybrids that are protected from DNase digestion precisely because of hybrid formation, neither the cellulose purification step nor the HPLC gold standard would be expected to remove them, and neither antibody assay at dot-blot mass loadings would reliably detect them at low abundance against the background of a multi-kb ssRNA preparation.
>End Claude.ai
The best way to eliminate RNA:DNA hybrids is to use a DNase that actually digests them before these purification steps. Something BioNtech was also fully aware of in the Lenk et al paper as they note Sutton et al documented that DNaseI digests RNA:DNA hybrids with 100 fold less efficiency than dsDNA alone. Many other nucleases exist that solve this problem (DNaseI-XT being just one of them).
Instead of solving this problem, they designed a qPCR assay that would never find it. We describe this qPCR game in the Journal of Independent Medicine. To this day, news agencies, fact checkers and even the New York Times recent plagiarism of Maryanne Demasi, PhD exclusive article on the ACIP meeting, claim this DNA contamination work has been debunked because regulators simply took the CT values from a single non-spike PCR amplicon from Pfizer and believed them. They never asked Pfizer why they wont report the CT values from the Spike qPCR assay they have listed in their EMA documents?
There is 100 fold more spike DNA in the shots than the non-spike DNA as the spike region has RNA:DNA hybrids that DNaseI cant digest. Pfizer knows this as Lenk et al is from Pfizer and published this 100 fold failure. These purification papers also underscore that they know this material survives their purification process. These authors are also part of BioNtech.
The only thing I can take from these disclosures is that Pfizer wants the RNA:DNA hybrids in the shots but they don’t want the regulators to know its there. Why would Pfizer want these additional inflammatory molecules in their shots? Do they not illicit the desired response without them? I will point you to substacks from Jessica Rose and Dr. Robert W. Malone to answer that question as it brings into question immune tolerance and perhaps the chronic disease market they now profit from with vaccine injuries: a class of injury that has escaped having an ICD10 code to track until very recently. That is in fact a very clever business model. Profit from a large injury market you created while the health care system has no capacity to track it back to you.
Yes, many of the vaccine injured patients are being given drugs made by Pfizer. Eliquis is just one anti-coagulant that has seen sharp increases in sales since 2020.
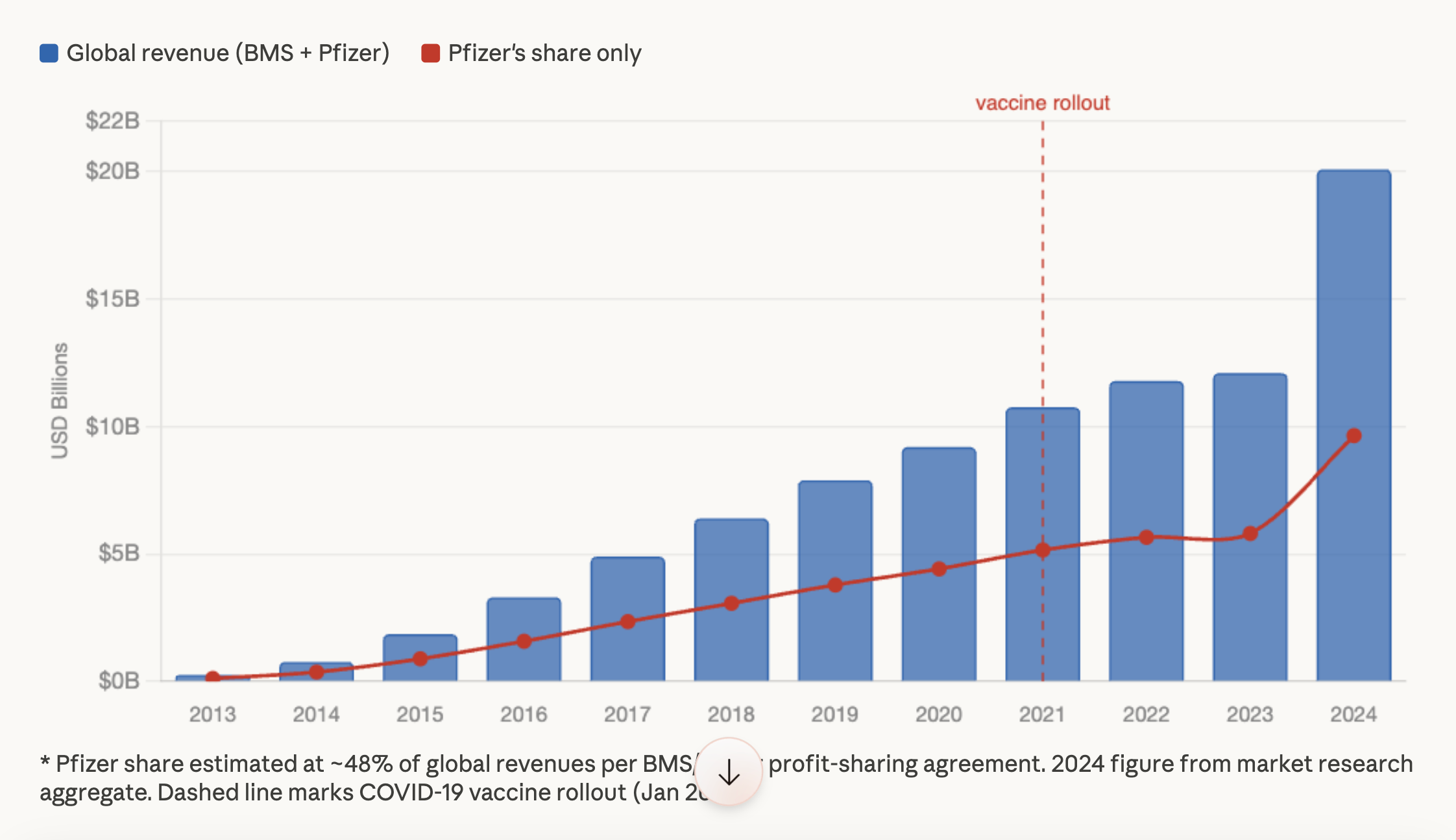
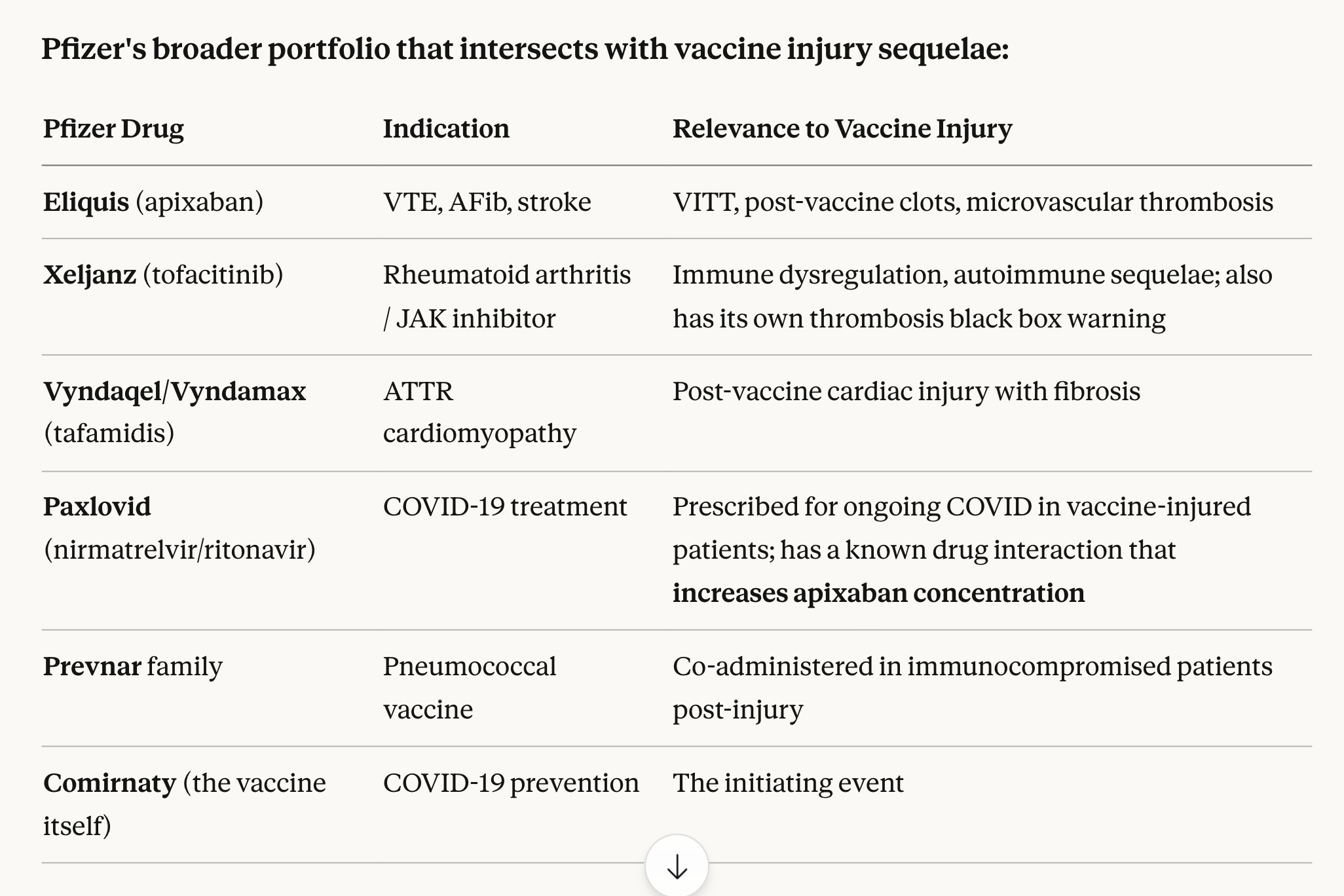
Well done Kevin!
I DON’T THINK THIS IS CUTTING THINGS TOO FINE:
Pharma accepted immunity that recognized potential for error
and presumably held companies harmless when committing these errors.
Engaging in procedures that are known to actually or potentially
cause death or long term harm is not error and should not be protected.