

Quasi-Species Swarms in SARs-CoV-2
How diverse are the swarms?


There has been a lot of discussion lately about SARs-CoV-2 existing as an ever evolving shapeshifting quasi-species swarm.
Some Background
JJ Couey has covered this and its worth entertaining. There are a few points I have contention with in his description of coronavirus conservation. About 1:27 into this cast he speaks to conserved protein regions in CVs which can provide cross reactive immunity. This is true but this doesn’t always translate into conserved DNA sequences as DNA has wobble bases that can diverge as silent mutations and often the conserved amino acids are not derived from the same DNA sequence. So I don’t think this is a fair evaluation of that Bat CV paper. This is also why taxonomy is always anchored in DNA sequence not Mass Spec of protein fragments.
There is second cast on this swarm topic from J.J as well which goes over a great presentation from Illumina on the nature of DNA sequencing technologies. 1 hour an 31 minutes in J.J begins to dissect this sequencing tech video and he highlights some details most of the public are unaware of in CV sequencing (amplification primer bias) which will be discussed below. He also gets a few things wrong but this is not his field and he is humble about his process of learning this on the fly.
The most glaring error I see is the assertion that these sequencers are akin to Theranos technology. For those who don’t recall the Theranos scandle, they never shipped a product. Illumina, Life Tech, Roche, BGI, PacBio and ONT have shipped thousands of DNA sequencers all over the world with 10,000-100,000s of researches using them, publishing with them and verifying each others work for nearly 2 decades now. It is hard to point to a more rock solid technology that exists in the life sciences today.
So what is this ‘Scoobie Doo’ Swarm Theory?
This swarm theory implies that SARs-CoV-2 did not come from a lab but was in fact always around and just emerged from the swarm. Over a year later it disappeared from the swarm with Omicrons introduction. Pharma, and politicians capitalized on it with sloppy PCR, rigged public dashboards, bought and sold MSM and controlled social media. Like many hypothesis there is 80% truth and 20% missing data we are just guessing at.
JJ has even suggested the Lab-Leak debate is a controlled opposition narrative attempting to distract the population from the fact that these swarms will always exist and at any moment governments can aim their PCR primers at a new member of the swarm and reinstitute lockdowns. As long as they can scare the population enough with the possibility of a ‘bioweapon leak’ from a political enemy or a new scariant, the population will accept the lock down and fight over how to better regulate labs and torture Fauci gremlins. This is what he refers to as being ‘Scoobie Doo’d’.
Early in the pandemic I made note of the fact that the government had such thorough control over the narrative and centralization of PCR, that they could have pulled this off with OC43. The only problem with OC43 is that it’s been circulating the globe for years, so unless there was something new about it, why worry? So ‘they’ simply needed the narrative of novelty to scare the population into compliance. The excess mortality barely demonstrates a pandemic of sizable proportion, or at least a pandemic that is not easy to disentangle from mandemics (iatrogenic effects) during 2020. These numbers are hard to reconcile given ACM (All Cause Mortality) rose after the government instituted vaccine mandates. There was a wave of excess death in the spring of 2020 mostly consisting of the comorbid and elderly. Much of this is hard to disambiguate from ventilator malfeasance, death certification fraud and other Mandemic artifacts.


So the magnitude of the C19 pandemic has certainly been exaggerated and this thread isn’t meant to resolve the % of death from the virus or from our misguided response to it. The thread is meant to expose uncomfortable data for the argument that the virus doesn’t exist. In science, ignoring uncomfortable data can often lead to bankruptcy.
Independent of the disease burden of C19, what we do know is that this RNA has been circulating the globe and has been mutating at a very defined rate. 6.5M genomes are now public in NCBI. This is decentralized data. Researchers from all over the world, using different methods are depositing data into NCBI and coming up with similar conclusions about the sequence of this RNA. Decentralized reproduction of results is the highest pillar of evidence we have in science.
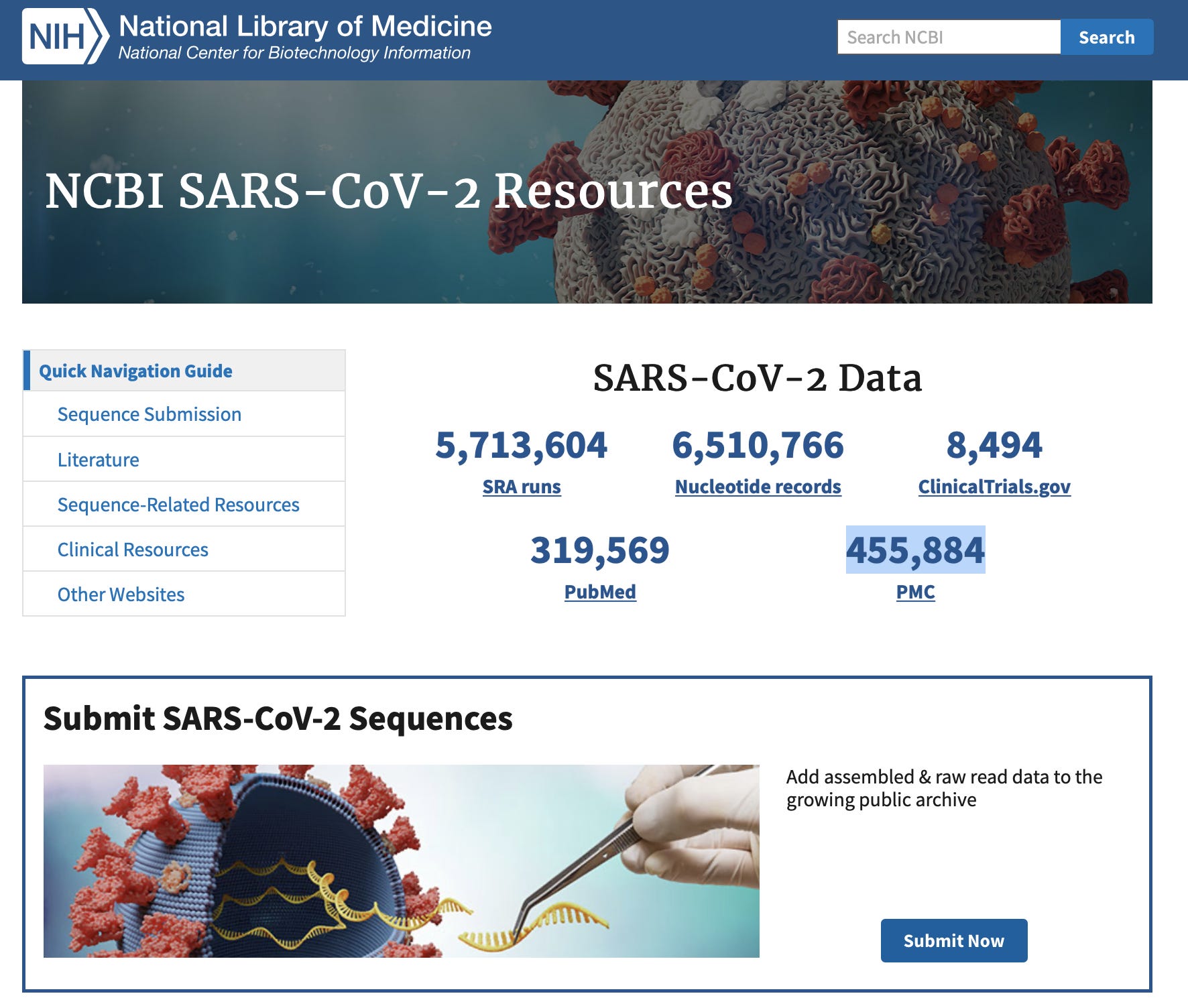
Primer Bias Concerns
Detractors of this data like to point out that many of these genomes are generated with PCR which will only sequence what you are looking for (SARs-CoV-2) and as a result this data is all an illusion reinforcing the ancestral virus and camouflaging the non-amplifiable divergent swarm. There is some truth to this as many submissions use the ARTIC PCR primers which amplify the 30kb genome with 2 pools of over-lapping primer pairs. This amounts ~99 primer pairs and you can buy them from IDT here.
Why so many amplicons? With polymorphic viruses you need redundant primer sets. In the event one primer falls on a variant, another primer will serve as a back up. This makes the process more swarm proof.
You will notice they are on V4.1 of these primers. That is because when the highly diverged Omicron appeared, 2 primer pairs out of ~99 dropped out and they needed to update them. So when suddenly diverged members of the swarm do emerge they are detected and corrected so the primer sets are still tracking the dominant RNA species being traced with qPCR. You might recall the TaqPath qPCR assay also had SGTF (S Gene Target Failure) with Omicron but the other 2 other primer pairs in the qPCR assay still picked it up. Single amplicon assays were notorious for this type of drop out and also had primers that picked up SARs1.
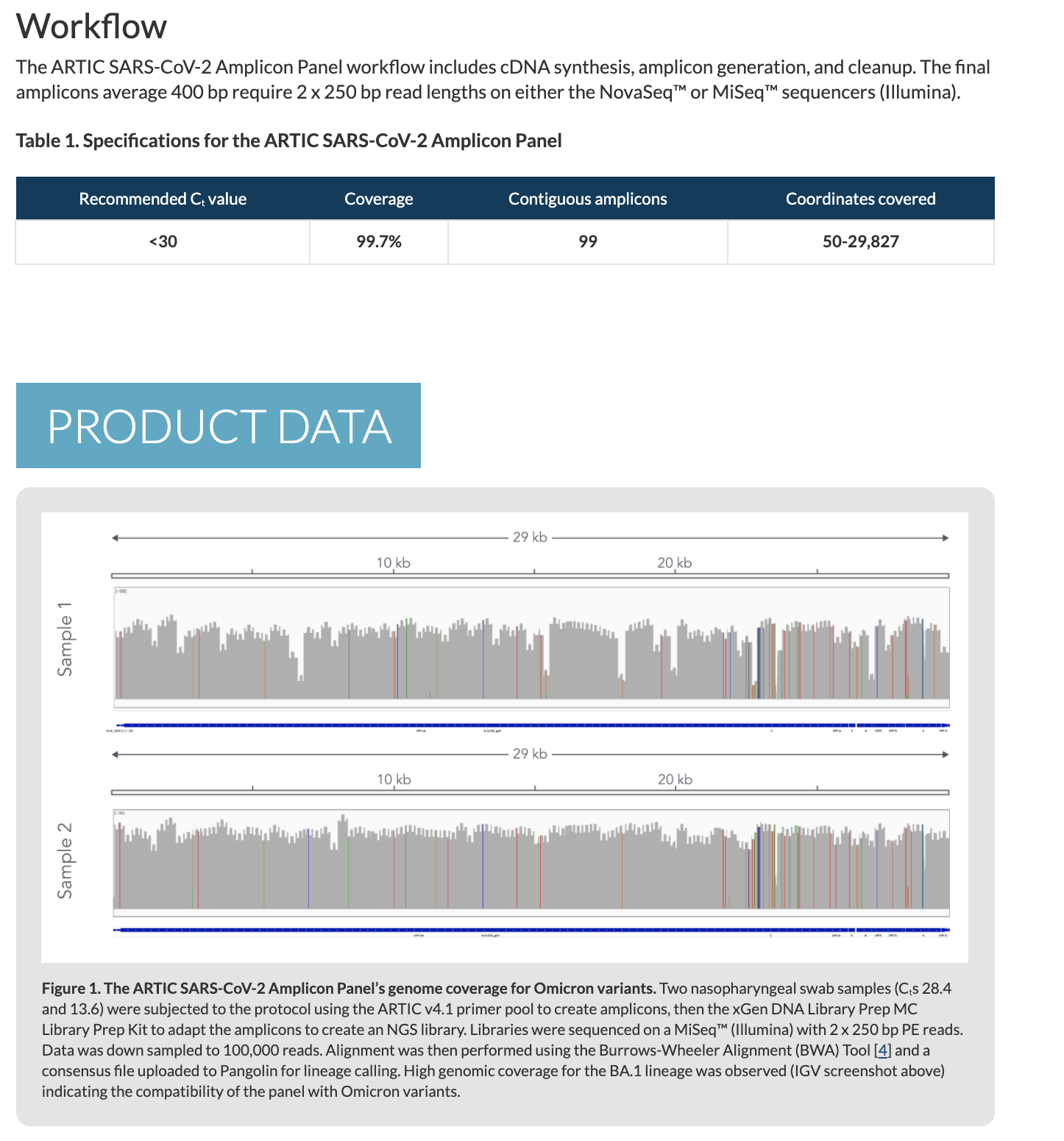
Non-PCR based approaches
Regardless of the stones thrown at the ARTIC process, what these arguments fail to realize is that some of the genomes in NCBI are derived from whole transcriptome sequencing. This is the process of taking all of the RNA in a patient sample and putting (ligating) universal primers on all of the molecules and amplifying and sequencing everything. This will include human, bacterial, fungal and viral RNA. It will miss DNA based viruses and potential circular lariats or viroids. Other methods can be used to tackle DNA based viruses but the below work focused on RNA based viruses.
Butler et al from Chris Masons lab takes this whole transcriptome approach and even compliment it with spatial transcriptomics on autopsy samples which helps to pinpoint where in the tissues the mRNAs are being expressed.
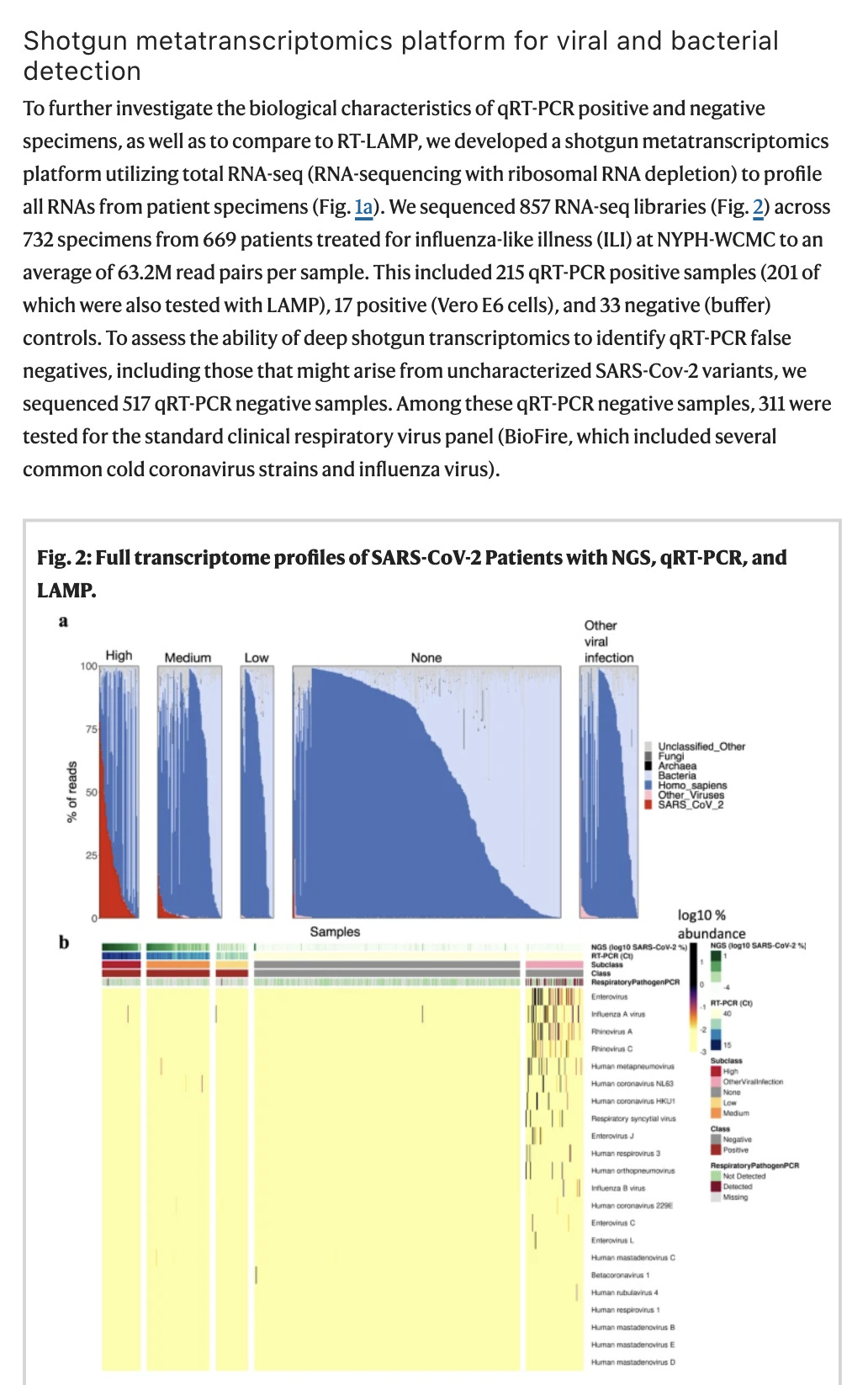
Most notable from this work is that by sequencing all RNA in patient samples, we can shed light on the blame game. Many people like to claim C19 is just a passenger and something else is causing illness. But that something else has no RNA signatures in Butler et al. Only 3.2% of the samples had co-infection with other RNA based viruses and in many patients the C19 RNA was the dominant RNA in the sample, even more prevalent than host cell RNA!!! This is consistent with Kim et al and Long at el.

They also get some information on the diversity of the swarm. Spoiler alert.. It’s not as diverse as other RNA viruses or plant and fungi infecting viroids. This is likely a result of SARs having an ExoN gene known to be 20X less error prone than other RNA viruses.
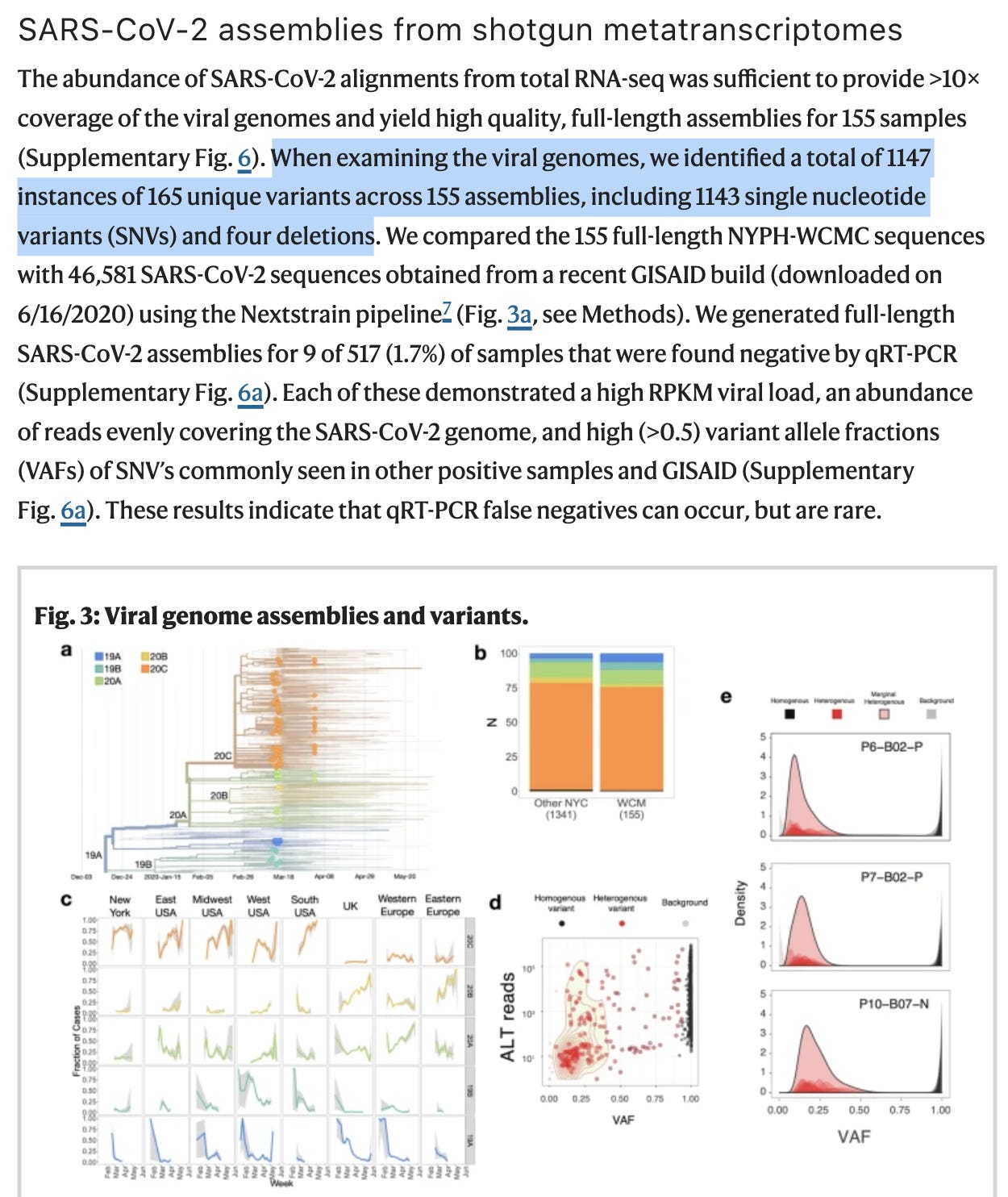
A close up on this swarm is seen in Figure 3d. Many variants are homoplastic (homogeneous meaning all RNA strands share the mutation) and a few are heterogeneous (Heteroplasmies where a fraction of the reads have the variant) with allele frequencies in the 5-95% range.
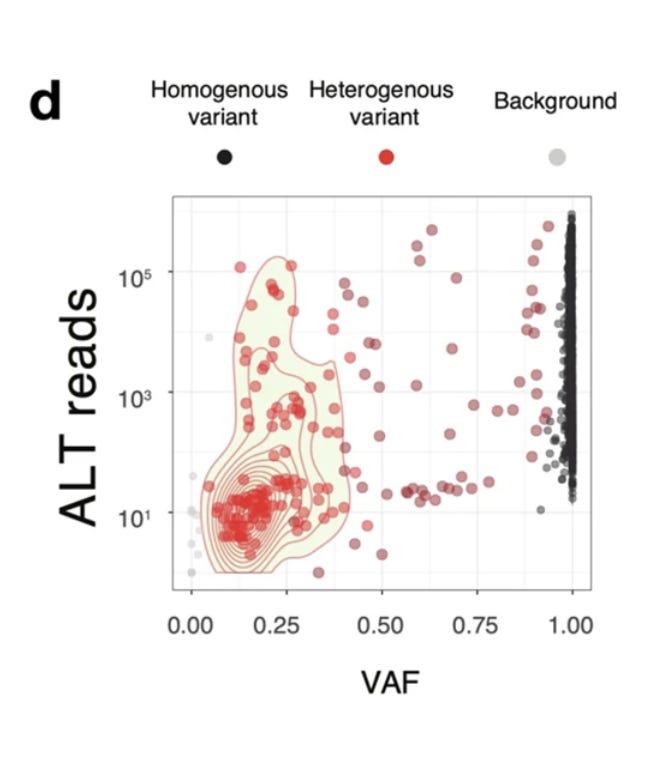
Another paper that attempts to dive deeper into low frequency variants (heteroplasmies) is Pathak et al. They are likely approaching the error of the cDNA synthesis step (1:50K). Even at these very rare allele frequencies they are only finding 11 strains per patient. I think this paper is likely conflating RT/cDNA synthesis errors with real APOBEC generated SNPs in the virus. Perhaps 1:1000 allele frequencies would be a more conservative frequency to measure as it would be well above the error floor of the sequencing reads derived from RT polymerases used for cDNA synthesis. I suspect this filtering would demonstrate 1-2 strains above the 0.1% frequency.

A good 20 minute video describing the sequencing assembly tools used to assemble these quasi-species swarms can be seen here.
Addressing the DNA Assembly Canard
Some other folks in the ‘Virus does not exist” camp tend to equate the process of DNA assembly to ‘Ferguson modeling’. This is far from true and is the most obvious tell that they are not fluent in sequencing technologies or the quality scores that are computed for each base and each alignment. Fergusons work is not predictive and based on speculation. DNA assembly algorithms are first benchmarked on synthetic data and later refined on real world data with known truth sets. 30Kb genomes are easy to assemble. Categorizing all of the subspecies, however, requires specialized assemblers like SAVAGE seen in the above video. Much of the sequencing surveillance is simply mapping to a reference genome and likely only seeing dominant members of the swarm with over 5% allele frequency.
But… There are technologies in the market that can read the entire gRNA in one read with no assembly required. So even if this DNA assembly Canard were true, it would be irrelevant based Kim et al.
Kim et al. used Oxford Nanopores to sequence the RNA directly without any conversion into cDNA. This platform sequenced the whole virus with 30Kb reads. No fragmentation and assembly were required. You can see a minumum of ~2,000X coverage across the whole virus in Figure 2B and 400,000X coverage on the 3’ end of the virus (right side of the virus). So lots of subgenomic RNA is made and a minority of the transcripts are full length. The paper further itemizes that middle region of 2,000x coverage and find 111 reads that span the entire viral genome.

The other detail discovered in this paper is that 65% of the mRNA in the sample is SARs-CoV-2 RNA. More viral RNA than host RNA! This supports Butler et al.

Long et al discusses studies that looked at the sgRNA versus the gRNA copy number. When cells are pelleted, 55% of the viral RNA is sgRNA. When RNA is extracted from the supernatent after pelleting cells, the number drops to 0.4%. This implies the large amount of sgRNA is unlikely to be packaged into virions as virions remain in the supernatent. sgRNA can still infect neighboring cells via a tight junctions but if it fails to package then it wont transmit via virions to other people. Therefor the sgRNA shouldn’t be considered part of the transmissible swarm.
One comment in JJs cast I feel needs correcting. At 1:47 he equates sequencing tech as a Therano mirage.
First, one does not need to accuse sequencing of being a Theranos mirage when the sequencing actually supports the existence of these swarms and actually quantitates them for you. The sequencing also supports the propagation of a what looks like a man modified FCS for the deadliest portion of the pandemic. Omicron has a significantly altered FCS site with critical mutations in the SEB domain and the regions flanked by BsaXI sites in the genome. If pre-omicron was a lab leak, waste water data and antibody data suggest it predates December 2019. It mutated significantly pre-Omicron until another less virulent member of the swarm replaced it but this mutation rate never ablated the FCS. Likewise, this process of swarm divergence was too slow to prevent the alpha FCS virus from global reach. If anyone leaks a more virulent infectious clone, there is still risk that the rate of CV polymorphism and divergence is too slow to stop global spread.
Suggesting the lab leak is a diversion tactic to ignore, I believe is shortsighted. We should not turn our attention away from the real risks this research presents. Given the large number of lab leaks throughout history, I do not think it is wise to ignore lab safety and GOF funding, regardless of where the origin story leads us. Coronaviruses can be readily synthesized in a month for under $10,000. We have the sequencing information to show that the rate of divergence in the swarm is too slow to fizzle out global spread of pathogenic variants.
The exact magnitude of these variants pathogenicity is open to a fair debate, but this is missing the bigger picture regarding the ease of synthesizing and potentially leaking more virulent ones. The sequencing data clearly shows the duration of each variants and how long this clade remained the dominant member of the swarm. I see no reason a more virulent leaked clone couldn’t last just as long and lap the globe.
There is now a $100B incentive for a Biotech company or state actor to create a new virus and profit from the vaccines or testing industries that will quickly reboot for the next leak. It is also pretty clear, that you can get away with such a crime as long as your interests can be aligned with the aggregation of more political power for individuals in regulatory control of these markets. The companies that funded the WIV were some of the first to develop tests for viruses that look a lot like the blueprints laid out in the DEFUSE proposal. They would like nothing more than for you to help them privatize their gains while socializing the risks upon the rest of us.
In summary, I think the risk of lab leaks is still very real and the swarming affect wont dilute this out fast enough to prevent global transmission. However, often times viruses trade off virulence for transmissibility (Mullers Ratchet) and perhaps such an isolated clone would fizzle before it goes global?
This is a risk we can’t afford to retrospectively regret underestimating.
You must also take note of the circularity of this argument. We know swarms exist because of exquisite sensitivity of next generation sequencing technology but the sequencing data is Theranos like technology?
Pick one.
Post Script-
It’s important to be transparent with my biases. I personally don’t subscribe to the notion that SARs-CoV-2 is just like any other Coronavirus in terms of its pathogenicity. In 2021, during the Delta wave, I watched this virus run through our household with family members all testing negative prior to infection on RAT and qPCR testing, only to become symptomatic and then test positive on 2 different bioassays (RAT and qPCR). This is the sickest I have ever become from a respiratory viruses and I was a C19 minimalist. I was not a psychosomatic hypochondriac Karen. I wasn’t vaccinated. The only time I was closer to death was with Rhabdomyolysis from a tick bite a few decades ago. With C19-delta, I had a 100-103F fever for over a week, low O2, loss of taste and smell, A-fib, aches and what felt like mitophagy. I lost 10-15 pounds. This also occurred a year later with my Brother who presumably got Omicron. We are not comorbid and under 50. My brother is a blackbelt, Wim Hof type of a health nut and this virus sacked him as well and brought out shingles. Three members of our immediate family were knocked out of the workplace for weeks due to this. I didn’t get my cardiac strength back for 1-2 months. This type of disruption is not reflected in any All Cause Mortality statistic so I don’t find IFR comparisons to other coronaviruses very compelling data to suggest this is just part of a natural swarm.
With all of the talk of herd medicine we have lost our focus on Personalized medicine. There are now known genetic predispositions to severe outcomes with C19. It is possible we fit into this group and are genetic outliers but these stories are not rare.
When I raise this anecdote with folks who don’t believe viruses are real, they will point to PCR being fake and the RAT test could be positive but could just be a coincidence to happen to correlate with the spread of this infectious agent around our household. When I forward these people papers like Kim et al and Butler et al which demonstrate the lack of other pathogenic mRNAs present in C19 patients and C19 RNA being 65% of the RNA in the cell such that it dominates even the hosts mRNA output, they go silent. Some knee jerk back to claiming it is never been perfectly isolated and parade around this God of the Gaps fallacy. Baric synthesized this viruses genome, free and pure of any other contaminating peptides and demonstrated it is replication competent upon transfection. It doesn’t get any more pure of an isolation than that.
Appendix
Mutation rates of CVs are dependent on RdRp polymerase fidelity and Host APOBEC enzyme activity.
Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting
A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping
Theoretical Analysis Reveals Cost and Benefit of Proofreading in Coronavirus Genome Replication
A few other comments on JJs cast. About an 1:41 minutes in he seems surprised the sequencing community is gunning to sequence a millions genomes. I hate to be the bearer of bad news but this has already happened. Just a single genome center sequenced a ~250K human genomes in 3.5 years. They don’t need the pandemic sample collection to achieve this end.
1:43 minutes in, he questions if we can really get off of Moore’s law or if there is a Theranos game going on. It is a remarkable graph and I encourage people to scrutinze the history here as it was a wild time in human innovation. The information he is missing is that genomics was in 9mm spacing (96 well plates) in 2000 and in 2006 MANY competing technologies emerged that shrank this to 1um spacing. I was involved in inventing and commercializing one of these methods known as SOLiD Sequencing and each instrument needed a $30K compute cluster to keep up with it.
The slope of that curve is really a reflection of free and competitive markets. Free and competitive markets quickly eliminate bullshit artists that exaggerate their claims. It is government sponsored fields that can carry off a fraud for a long time and despite this presenter giving accolades to Francis Collins public human genome project instigating this technological revolution, nothing could be further from the truth. The sequencing companies that took the least government grant money dominated the market place (Illumina). The size of NHGRI sequencing grants issued to these next gen sequencing start ups is INVERSELY correlated to their acquired market share. That is an uncomfortable fact they don’t like to talk about that I will reserve for later article.
Also recall the circularity of the argument. The only reason we know these swarms exist is because of exquisite fidelity of Next Gen sequencing and now he’s tossing sequencing fidelity under the buss because for some piece of his argument he needs it to not be true.
JJ is dead right about Scott Gottlieb being on the Board of Illumina and was pimping public policy that increased sequencing demand. I cant fault Illumina for this as the mainstream media never stopped once to question these conflicts.
At 1:46 he mentioned we need clones to sequence. That was true for Sanger Sequencing but we became clone free with Next Generation sequencing. The way clonality is achieved in next gen is not through putting a single plasmid into a single E.coli cell and amplifying the plasmid through growth of E.coli. We resort single molecule PCR to amplify DNA in highly parallelized compartmentalization. Emulsion PCR or Bridge PCR made cloning obsolete for next gen sequencing. The 3rd gen systems that can sequence without any PCR just sequence molecules directly. This is a very important advancement as JJ is correct to point out that there was a replication fidelity loss with PCR. PCR tends to have an error every 10,000-100,000 bases. Plasmid are 10-100X higher fidelity but for the purposes of tracking swarms PCR is sufficient. RT-PCR is more error prone and closer to 1 error every 10-50k bases due to the lack of fidelity of RNA polymerases (See attached papers above in bold).
This leads into JJ thinking there is very little RNA, therefor it must be hard to clone. As the papers above show, C19 is the most abundant RNA in a cell and there are multiple sequencing platforms that have demonstrated sequencing these with ease. This conversation devolves into the primer bias arguments already addressed above.
All in all, this is an important topic and Im glad JJ raised the flag here.
I have forwarded to JJ.
I also want to emphasize that JJ is not a “No virus person”
He has also pointed out/asked why there are only 111 full length reads in the nanopore data KIm et al.
That is likely due to the fact that nanopores selectively load smaller fragments.
For people to get these glorious long reads on nanopores, you have perform all types of size selections to get rid of the small shrapnel otherwise they consume all the pore space.
I should have better explained that oddity in KIm et al.
I still think Couey's instincts regarding the meta are valuable - because, well, lots of people have already been making those same points, myself included: "lab leak" plays more like a deliberately staged counter-narrative, made to appear like a forbidden secret that would finally be "vindicated" in the mainstream so we could all go back to bed.
And the case for DEFUSE->WIV->SARS-CoV-2 is weak. WIV was focusing on southwest china derived genes, not northwest Laos (BANAL). And their style was way messier than the proposed SARS-CoV-2 BsmBI / BsaI map. So they aren't where it came from. Evidence continues to point to October 2019 as emergence (https://www.biorxiv.org/content/10.1101/2022.12.04.519037v1
), Wuhan was not NOT having the military games at the time.
I'm ambivalent about the case for SARS-CoV-2 really having "global transmission competence." Yes, you have FCS fidelity in all the clades, but said clades keep dying out, requiring these miraculous "resets" from B.1.1 backbones every year. Hmm. At best you could say BA.2 and 5 have finally demonstrated staying power.