Veterans Day Vials
5283bp read in very recent Japanese Pfizer lot.

I had the pleasure of training more excellent people on how to run our Spike/Ori/SV40 qPCR assays and in the process teach them a few QC’s that are helpful to troubleshoot these methods. First order of business was to put them (Charles Rixey and Jessica Rose) to work for MGC making some E.coli and Salmonella genomes:) We like generous labor and they were a pleasure to work with. Jessica even voluntarily cleaned our Kitchen when weren’t looking. Many great scientists have this type of OCD which is always a sign of meticulous bench practices.
MGC had Veterans day off so it was a great time to get access to the lab without any interruptions or distractions to the team. These were long days. Both folks had to travel some distance by train. Early days and late nights getting all this data together.
Jessica posted a video on some of this work which received over 500K views on X and evoked the typical swarm of chaos agents instantly making videos of their own chastising the work. We actually have a library of 22 videos as we filmed as much of the workflow as possible and we will be releasing those over the next month to help educate the world on how to reproduce this work and improve on our methods.
One of these videos caught the attention of multiple ACIP committee members as it nicely demonstrated the Mechanism of Action behind why the regulators are getting lower DNA measurements than everyone else.
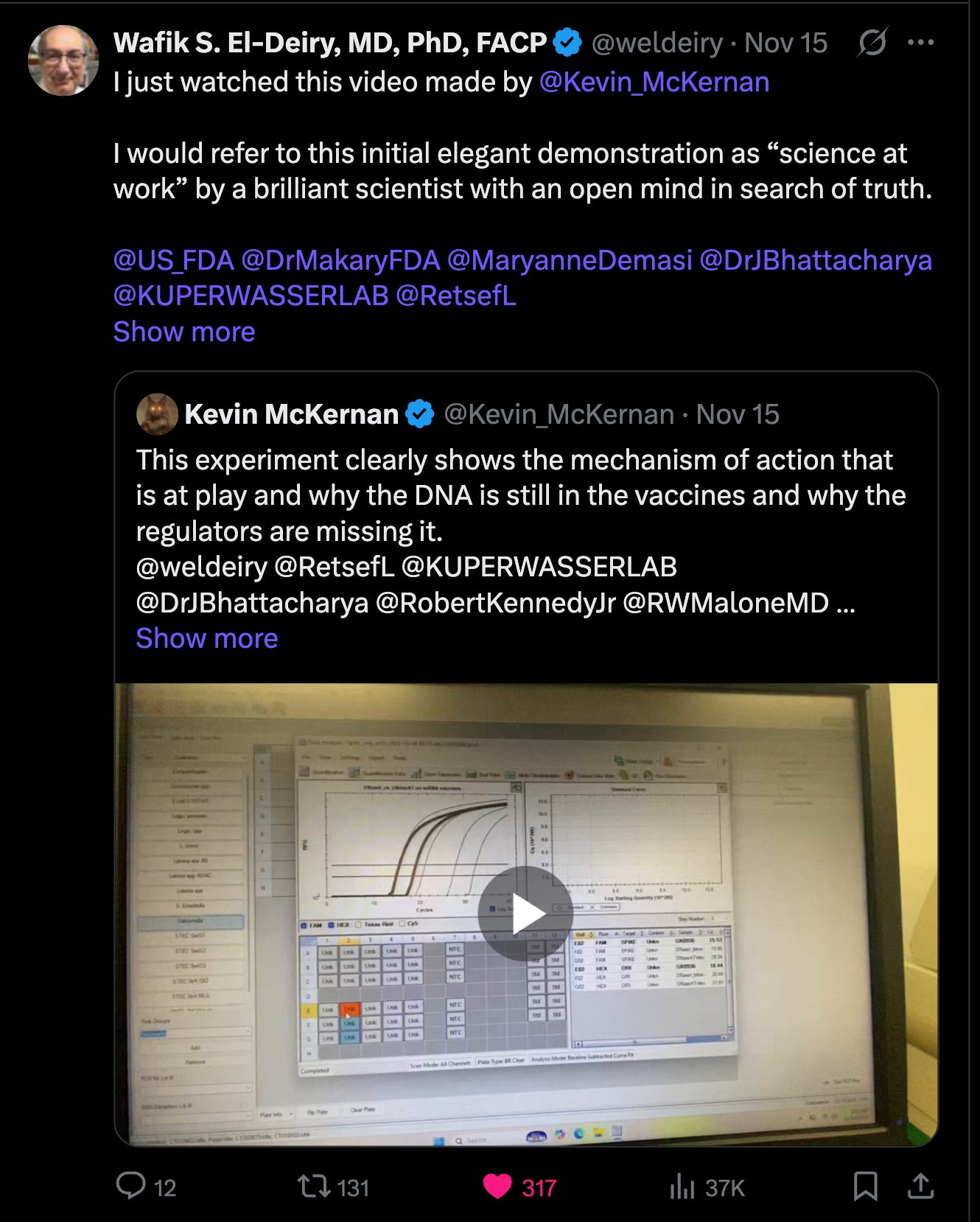

Jessica Rose covered this topic already so I will keep it brief.

The reason the regulators cant find the DNA is
1)They are not looking for it themselves and simply trusting Pfizer’s provided numbers (exception might be the TGA who claims to have looked at the KAN gene using Pfizers primers)
2)When Pharma measures this, they intentionally choose to quantitate the region of the plasmid that is easily destroyed by DNaseI and avoid at all costs ever looking at the part of the plasmid that cannot be destroyed (Spike). DNaseI cannot process RNA:DNA hybrids so the Spike DNA remains at 10-100X higher levels than the KAN gene.
This is very sus behavior as Pfizer disclosed a RT-qPCR assay that targets spike in their EMA documents.
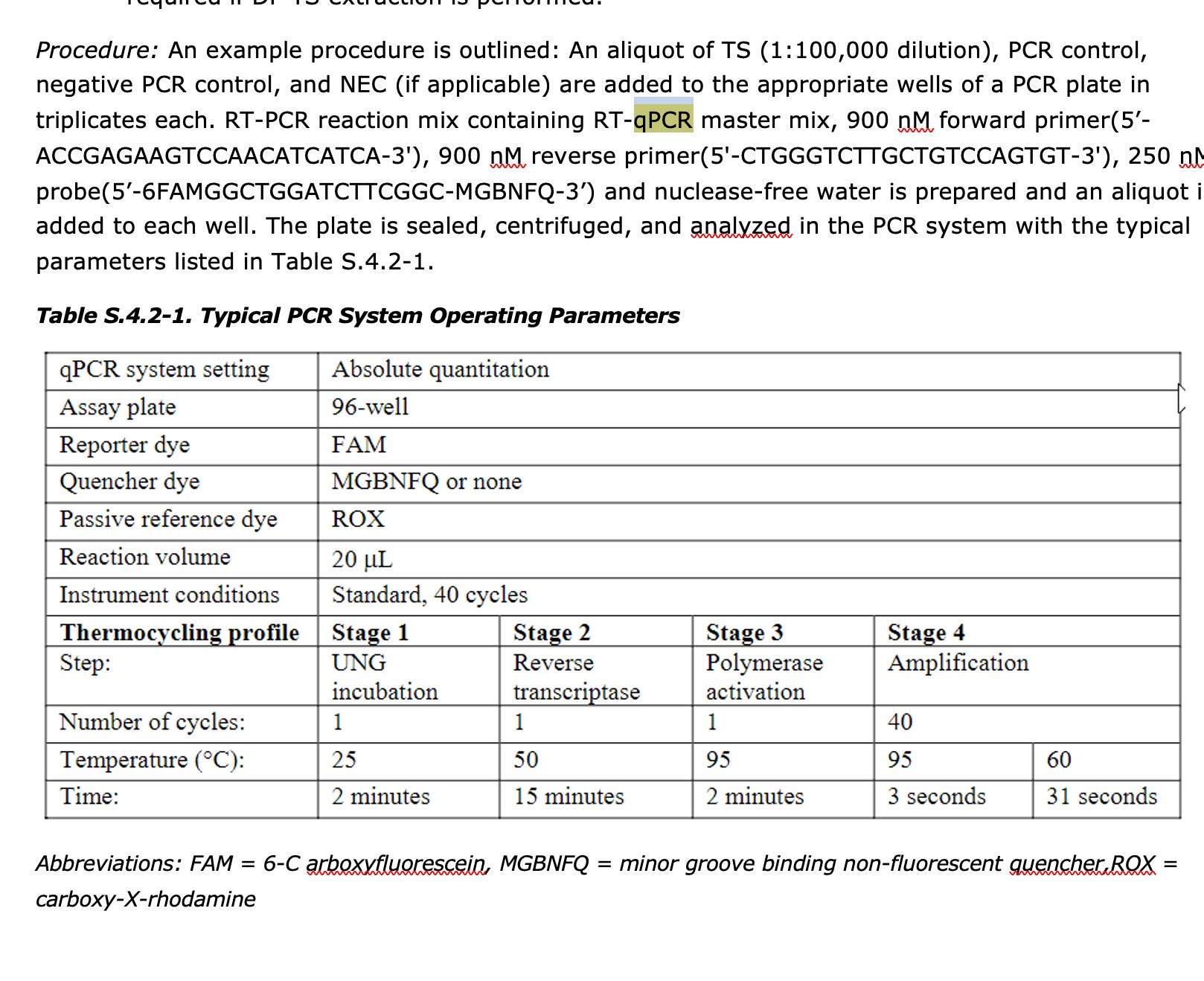
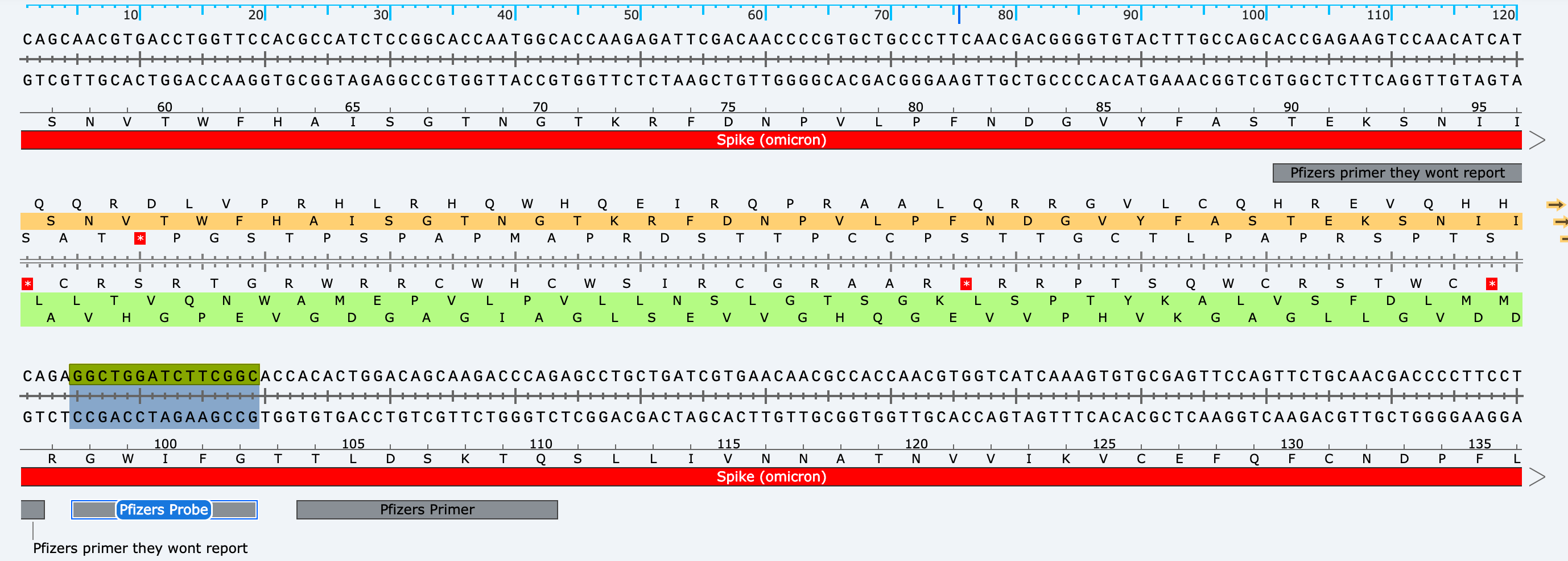
These primers land in Spike but they won’t ever report the quantitation they get from them. All that is required is to use a polymerase that doesn’t have RT activity and this assay will measure spike DNA in the product (not RNA). They will NEVER do this as it will reveal what everyone else is reporting.

What is very interesting about this exchange with the ACIP members is that it led to Senator Tom Corbin of South Carolina asking for me to testify about this topic in the SC Senate.
During that Presentation, Andrew Recknagel (from the Buckhaults lab) presented some of their data which recapitulated the same observation but with even more qPCR assay density (8 assays) than our 3 assays. Below is Andrews data on the copies of DNA reported from multiple assays including the KAN gene.
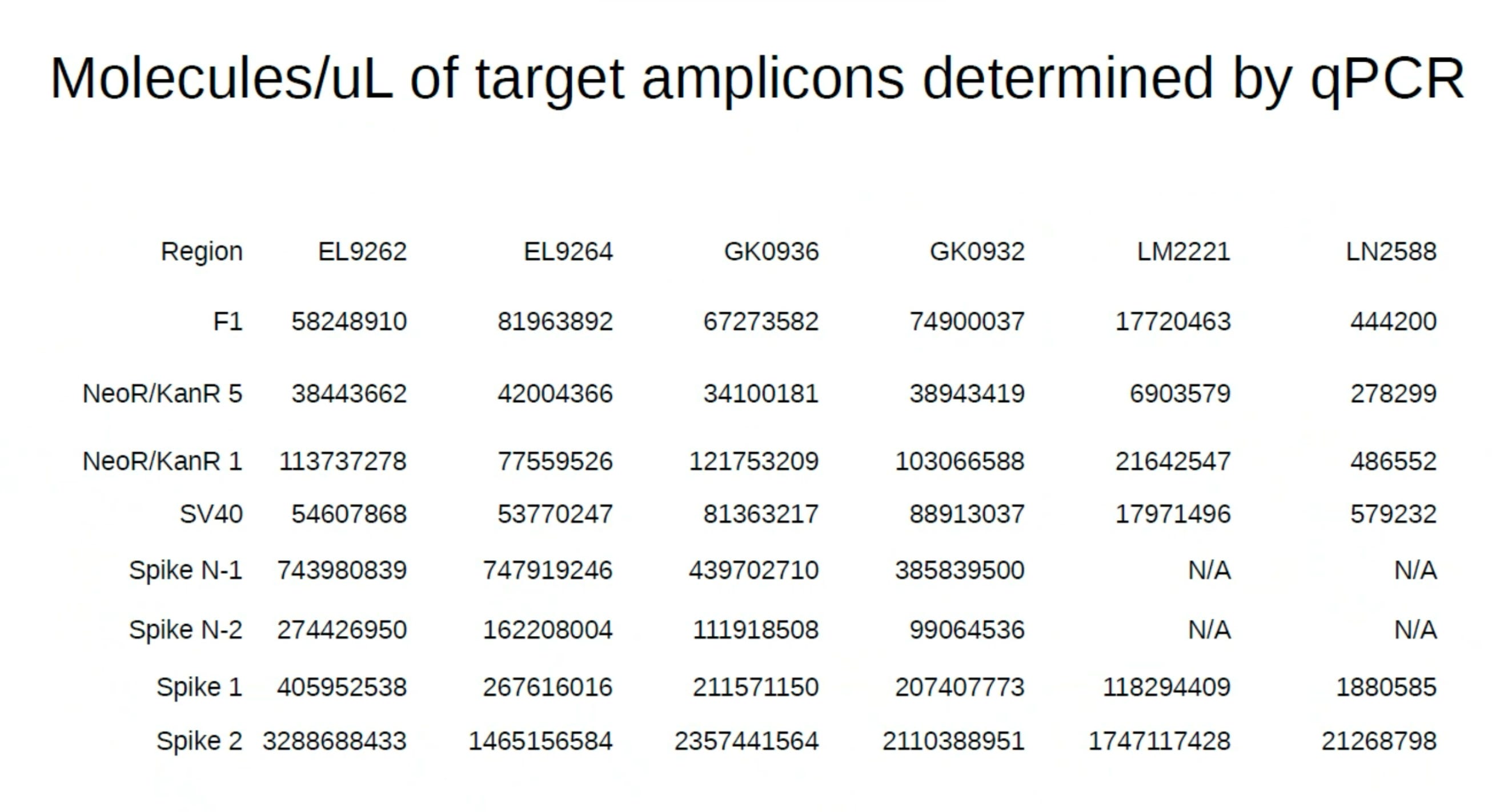
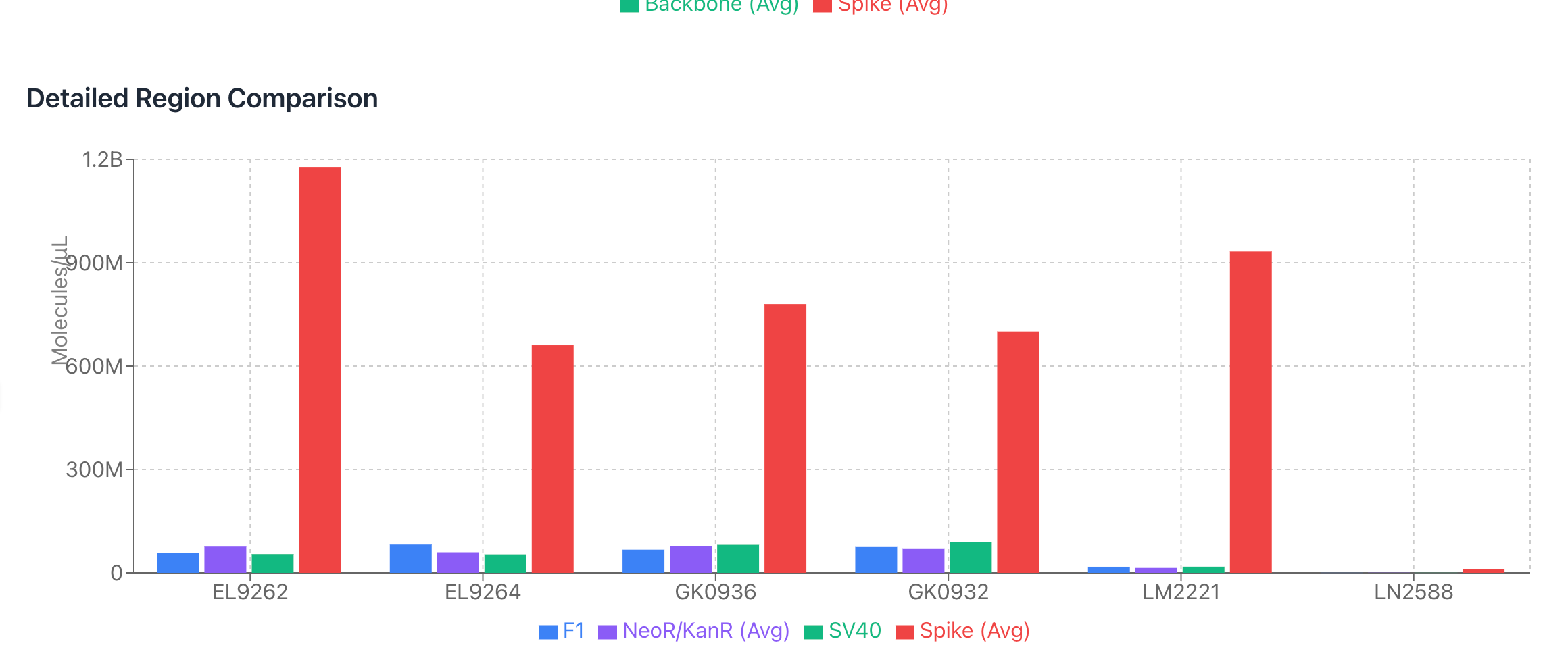
BANG! Same damn observation produced independently and with zero communication from us on what we were seeing.
Even Claude understands the scam. Using a single assay like the KAN assay reported by the TGA is a game of “Hide the Ball”.
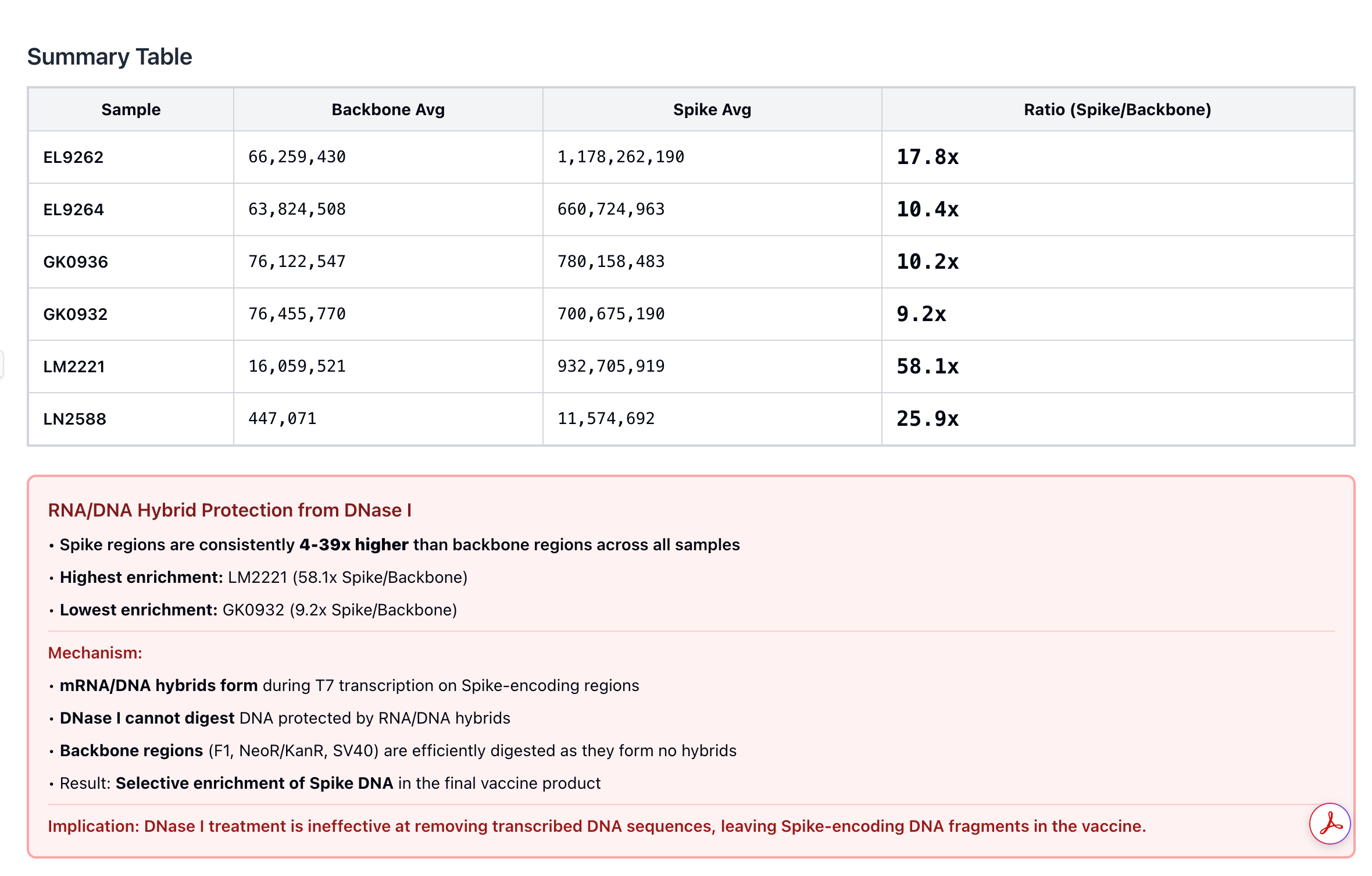
Andrews qPCR data shows Early CTs for some of the same lots we are handling (LN2588 and GK0936)
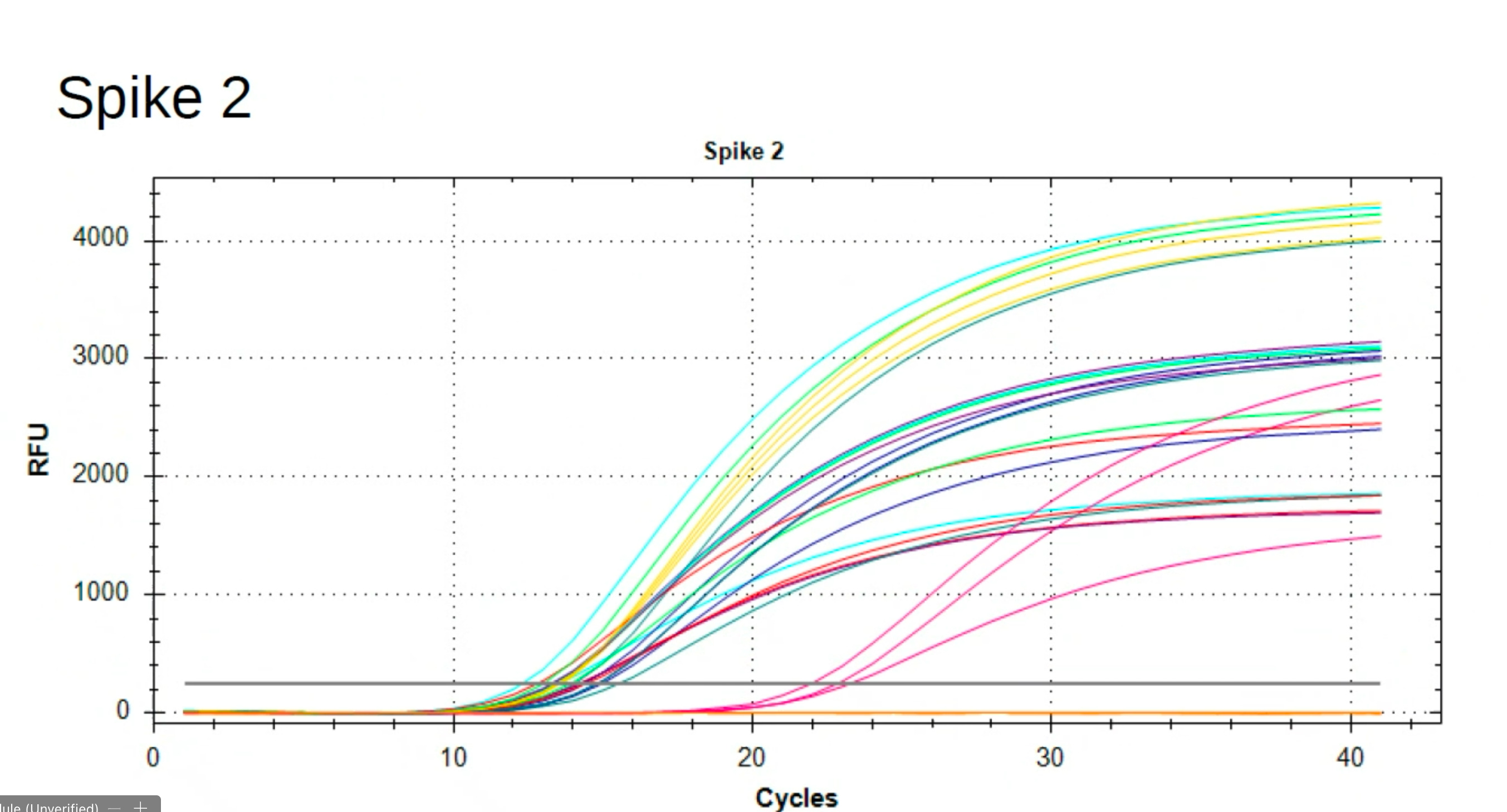
Sequence validation of these findings.
Some data for transparency purposes-
For these Oxford Nanopore runs, we 1st ran MGCs samples (E.coli and Sal genomes) as a practice run for Charles and Jessica but also as a means to save cash. ONT published methods that enable you to recycle used flow cells using an undisclosed DNase so when MGC finishes a run, we can wash their flow cells with a DNase and make use of the end of the arrays lifespan. We won’t get the most data doing this but we’ll get more than enough to answer some vaccine questions.
After this E.coli/Sal run, we washed the flow cell and ran Charles Library first.
1)Charles Rixeys Pfizer Vial Run for LN2588 and GK0936 (22 files, 23Mb).
After 2 hours of running we spiked in Jessica Rose’s Moderna vials which had far lower DNA contamination (not related to her) and thus less productive read generation.
3)We rebooted the computer and reran this library overnight.
4)Nearly a week later when more Flow Cell space opened up we ran the E-Lots (BAM files). Fastq files.
The 4th run of vials (E-Lots) were previously opened, we have only 100-200ul in each vial and had to DNA prep them independently and then pool them for ONT library construction.
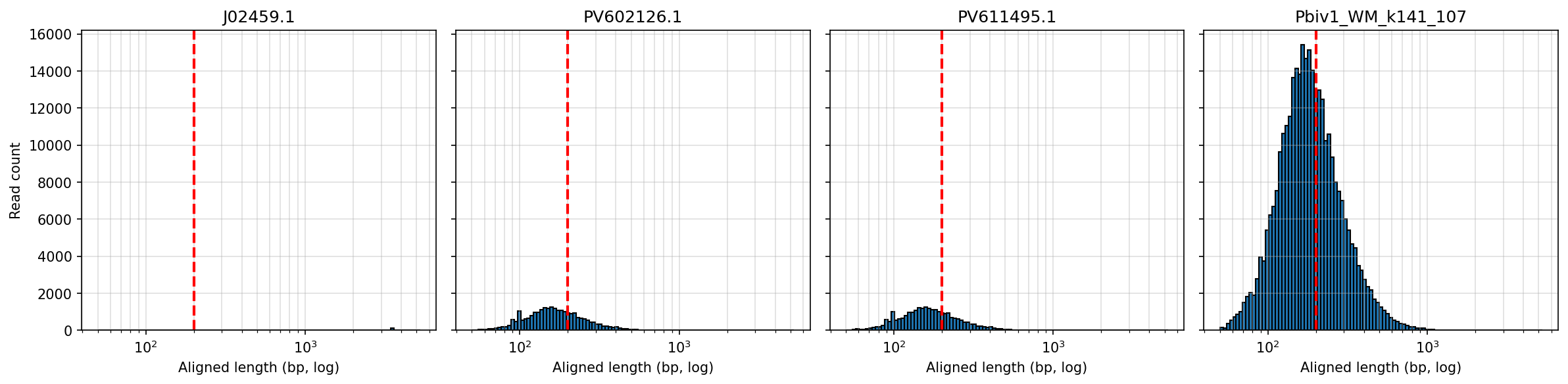
So there are 4 batches of reads above and its important to note that data set #2 has Pfizer and Moderna sequences pooled together that we can sort out after the fact by mapping them to their respective genomes. Each library had a 1ul spike in Positive control DNA (Lambda).
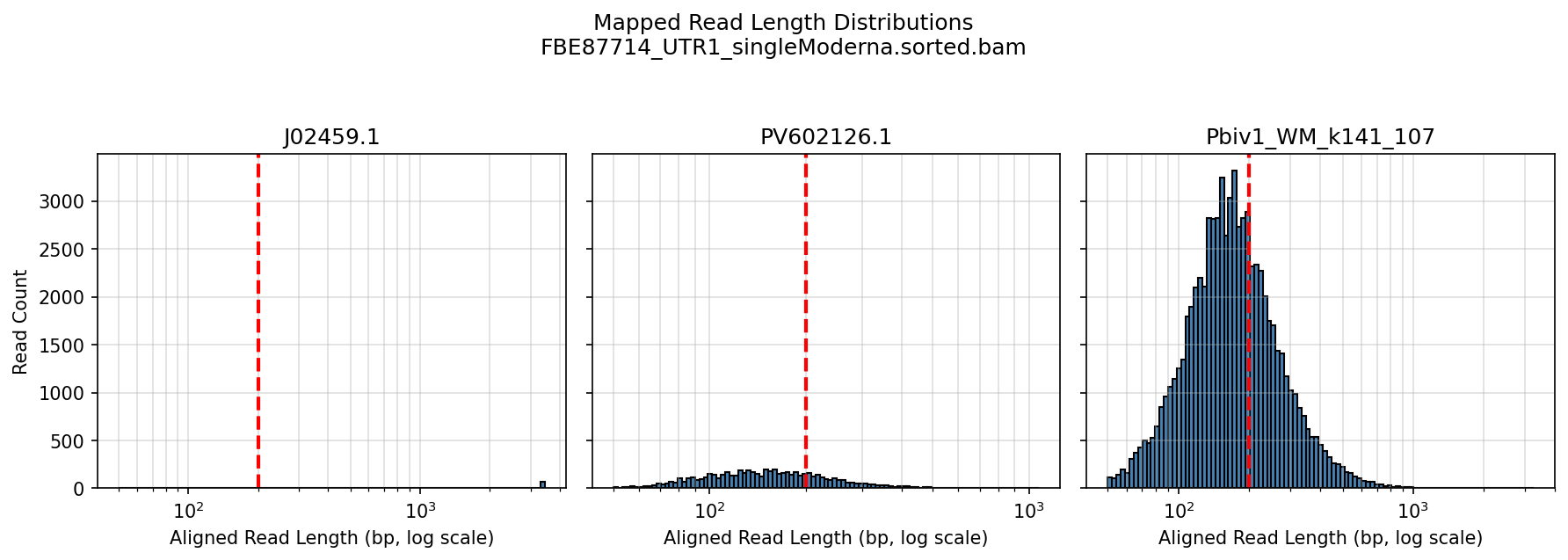
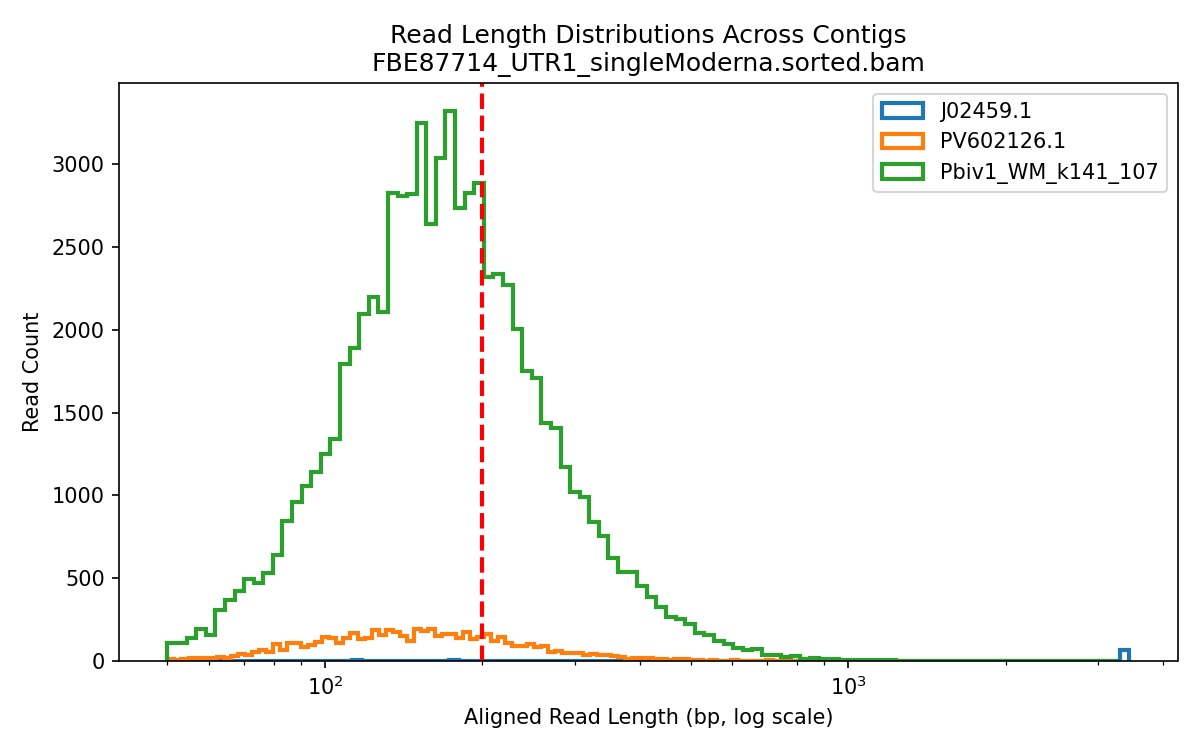
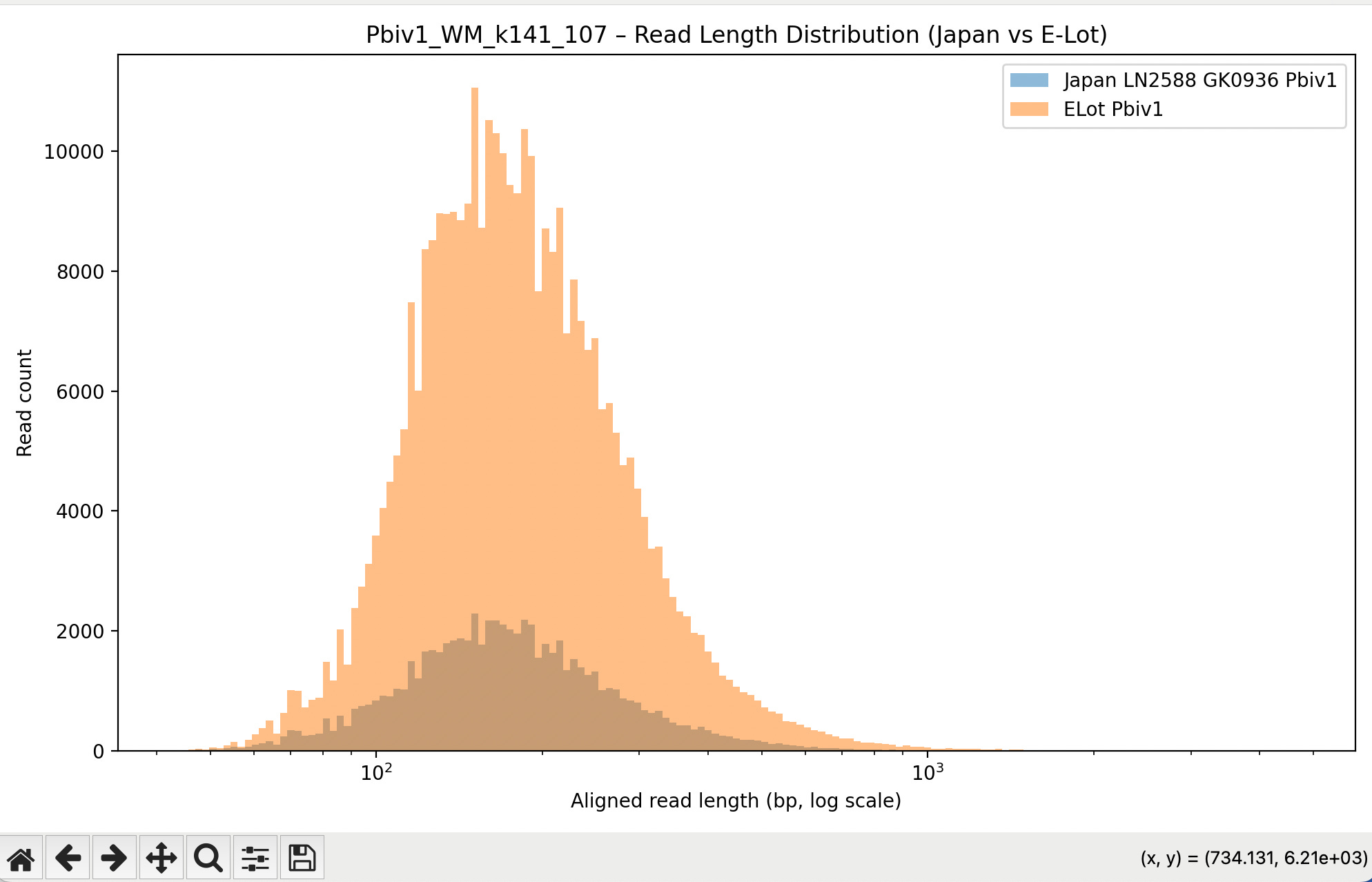
Below is the aligned length of the reads from the 1ul Lambda positive control sequence (Left), The Moderna samples (middle) and the Pfizer vaccines (Right). The Red line is the FDA/WHO size cutoff.
The FDA/WHO don’t consider anything under 200bp to be meaningful because they are still stuck in the past where DNA was injected naked. The host nucleases destroy this stuff in 10 minutes. Once this DNA is in a Lipid Nanoparticle, it is protected from this destruction and is actually a higher risk for integration.
This video covers some Qubit standard set up and Oxford Nanopore Rapid Library construction. This was for the E.coli and Salmonella practice genomes.
We ran these 1st as they are faster and easier than the Ligation kits we use for Vaccines. If you need to maintain the fragment length, you have to use the Ligation library construction kits. If you have High Molecular Weight libraries and you can afford to fragment them into 3Kb libraries, the Rapid kits take half the time and work to use.
A few steps we modify when using the ONT ligation kits for Vaccines.
1)The End Repair step we extend to 20 minutes. This is needed as the DNA is fragmented and you have more ends to repair.
2)The 60ul SPRI step (1X volume) we extend to 90ul SPRI (1.5X volume). This is required to capture more of the small DNA. While the sequencer will have low quality on these reads, the following ligation step is very sensitive to being under fed with DNA. If the DNA concentration is too low, the adaptors ligate to each other.
3)The Ligation step is extended to 30 minutes. 50ng of 200bp DNA has 100X more ends to ligation than 50ng of 20Kb DNA, hence more ligation time is required.
4)The 40ul SPRI step (0.4X) volume is increased to an 80ul SPRI step (0.8X Ampure).
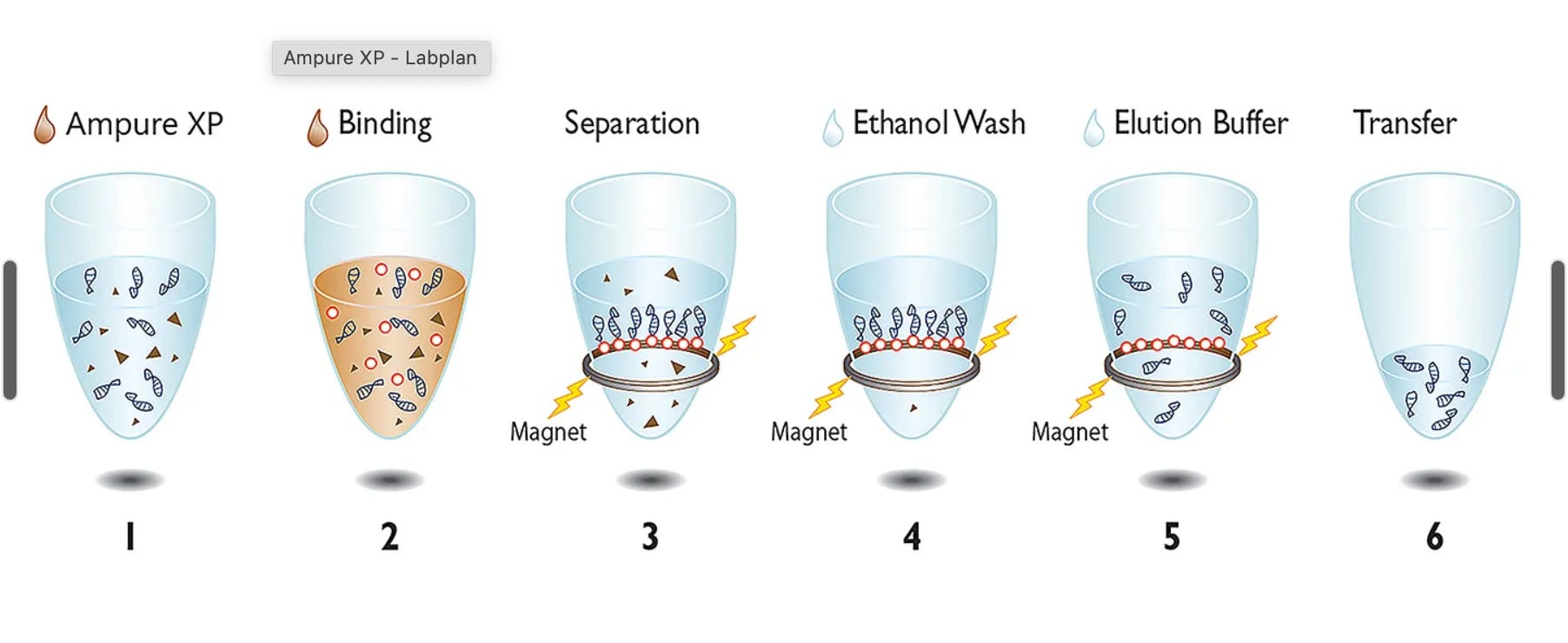
What are SPRI steps? This is a magnetic bead purification system that is very open source. You can easily adjust the target size of DNA you want to capture by simply changing the volume of reagent you add. Traditionally, with column preps, you need to use different columns to make this happen. With SPRI. you simply adjust the volume of reagent you are using.
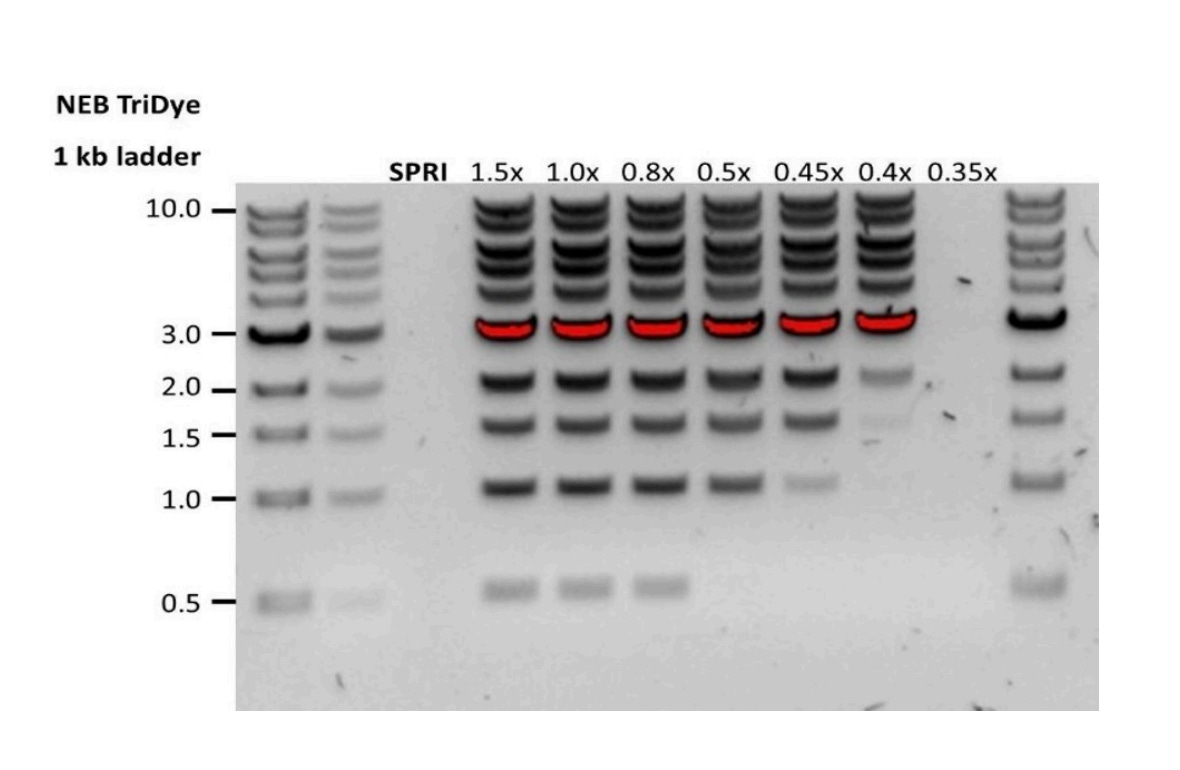
Below is an example of a 100bp ladder being purified with various volumes of SPRI. You can see if you add 0.4X Ampure, you are on the edge of loosing everything under 1Kb.
If you think the majority of your DNA is around 200bp, you need more like 0.8X SPRI to capture it.

Since Oxford aims for long reads, they regenerated this data with a larger DNA ladder. At 0.4X SPRI, you cut off most molecules under 2Kb. If we followed this protocol directly, we’d have very little DNA left to load onto the nanopores.
The overnight Rixey run has produced a record breaking read from the Pfizer vials. 5283 bases long. When the reads get this long you really have to map the reads to a concatenated reference to ensure you are capturing all of the reads that might span the linearization junction. This read looks like it maps right up to the Eam1104i site.

The below aligned read length histograms are from a library that had no Moderna DNA present. We still mapped the Pfizer reads to all 4 references. You will note some short fragments mapping to Moderna because short sections of Moderna’s plasmid are identical to Pfizers but zero reads over 1000bp bases map to Moderna.
Reads over 500bp
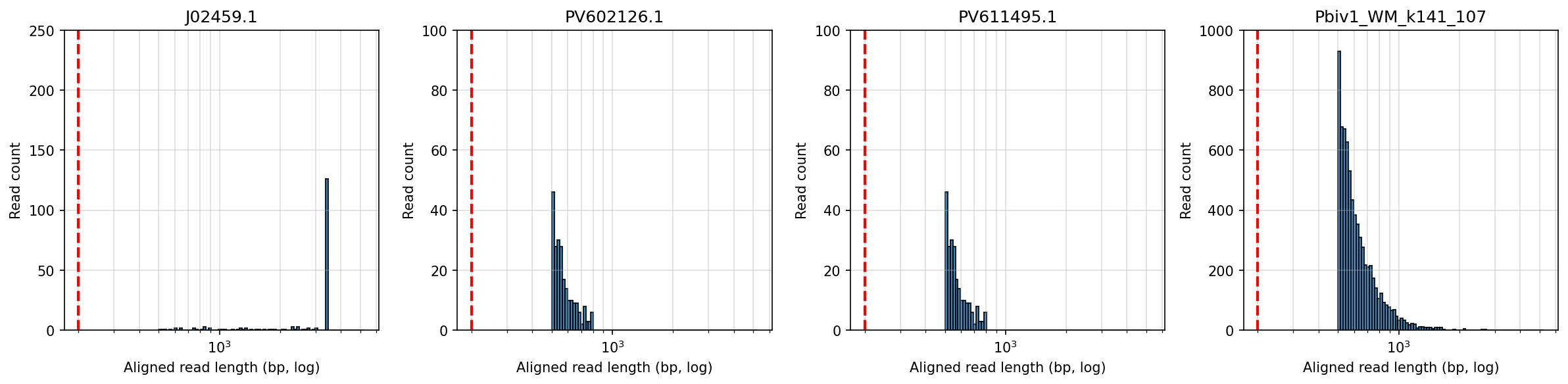
Reads over 1000bp (none for Moderna)
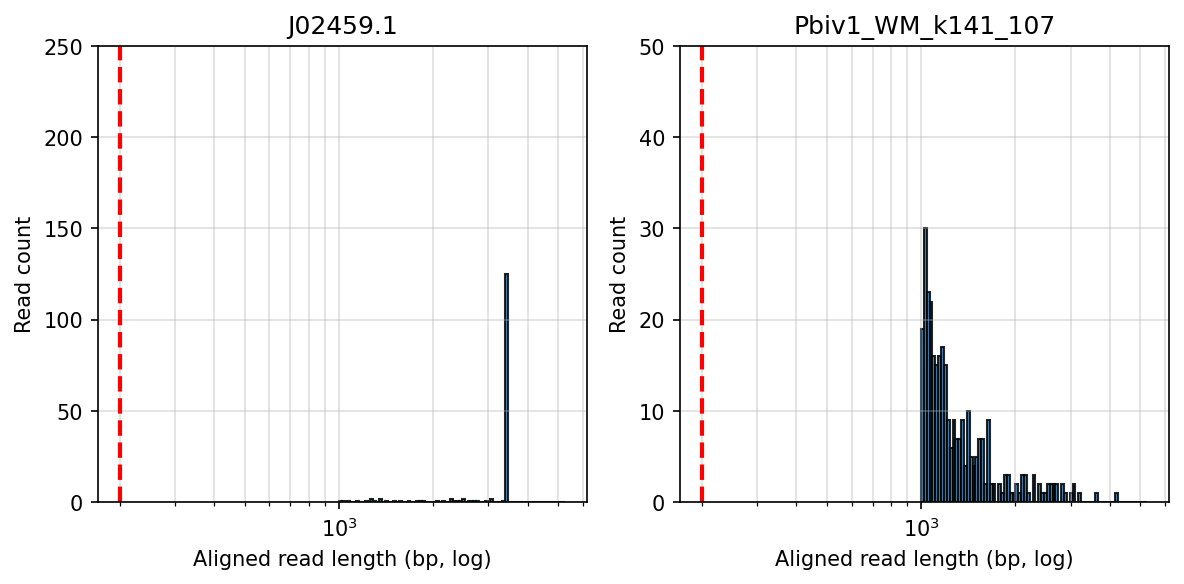
We then compared these Japanese lots to some E-Lots we were sent. We only had residuals on these lots so we had to pool them together to get enough DNA for sequencing. GJ6669, EW0196, ER8733, EW0178

Understanding Duplex Reads on ONT.
Here is one of the longer reads we found in the E-Lots.
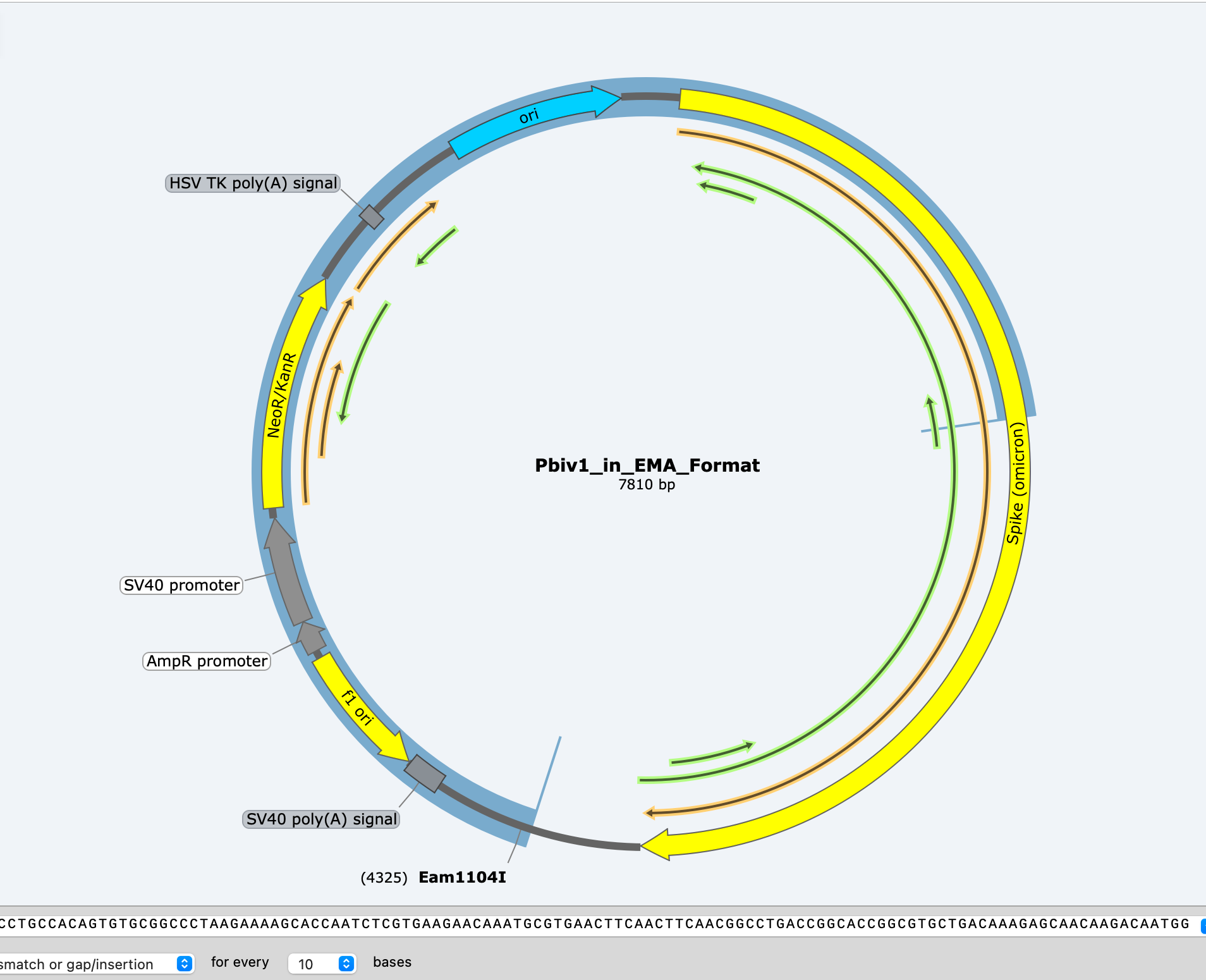
BLAST analysis gives what appears to be a 7810bp hit suggesting a full length plasmid!!!! But you will notice a break in the alignment in the middle (3000-4500). That is signifying that the alignment swaps direction at that point and heads back the other direction.

If you take the read and put it into SNAPgene, you can see the read is a mirror image of itself.
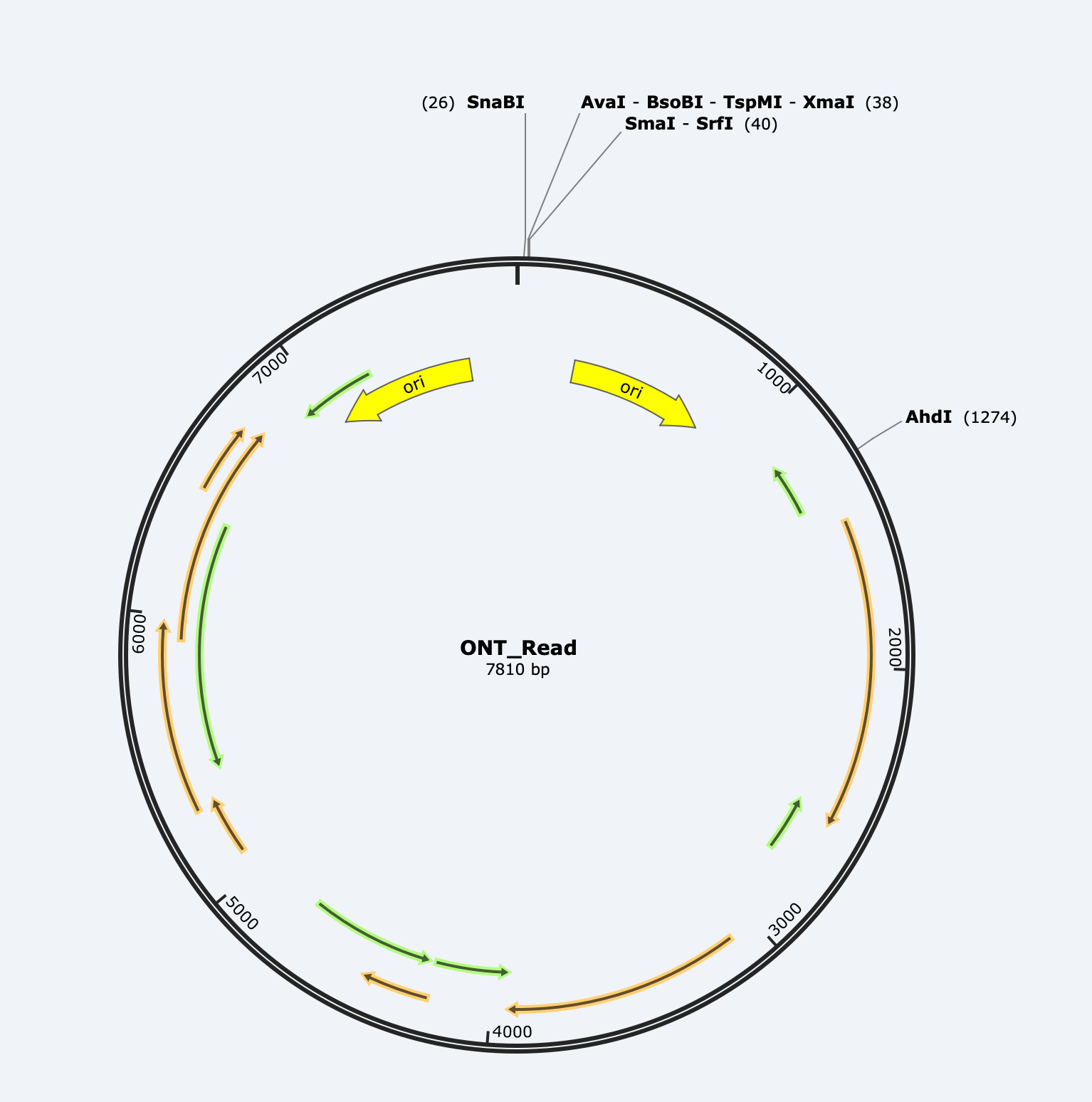
This is one of the features of ONT sequencing known as Duplex reading. The most likely DNA molecule to be sequenced after the 1st strand goes through is the opposite strand that was peeled away with the motor protein. This can help build accuracy as the sequencer sequences one strand and its reverse complement next. An excellent error correction mechanism. Occassionally, these duplex reads end up as one read in the same Fasta file as the time gap between strand 1 and strand 2 was too short for the sequencer to put them into separate files

Stay tuned,
We have much more data being crunched and will report back shortly as this all matures.