

Dirty Deeds Done Dirt Cheap
or maybe my expectations are too high?

I keep seeing data from various groups trying to look at this DNA contamination problem and I’m beginning to realize, not every molecular biologist spent their career designing nucleic acid isolation kits nor even specialized in plasmid purification technology.
There is a very specific reason why we used heat and Triton-X to evaluate the DNA inside these LNPs. It was rooted in my understanding that most DNA purification kits on the market are designed to eliminate small DNA.
In fact, this is often the number one feature. You want to capture the product of an amplification reaction (usually over 100bp in size) but thoroughly clean out the smaller input primers and nucleotides. This ability to size select DNA often makes or breaks a technology as a failure to remove the small excess primers and nucleotides will feed side reactions if you don’t remove them for later steps.
Have a look at Ampure. This is the technology we developed for the Human Genome Project. The salient feature that allowed Ampure to take over 80% of the next generation sequencing market is that its size selection was tunable. You could make it bind larger or smaller DNAs just by changing how much volume of the reagent you used.
This was not doable with columns which were all tailored to a pre-designed molecular weight cut off.
So if you want to purify 300-1kb PCR products you will need a different column than what is used for plasmids (1-10kb).
With PCR product purification, you need to remove the residual 20bp primers. Failure to do so will poison your Sanger sequencing reaction.
For plasmid preps, you have cut off everything under 500bp. If you use a PCR purification column on plasmids, you’ll get lots of small RNA and shrapnel DNA coming through the prep.
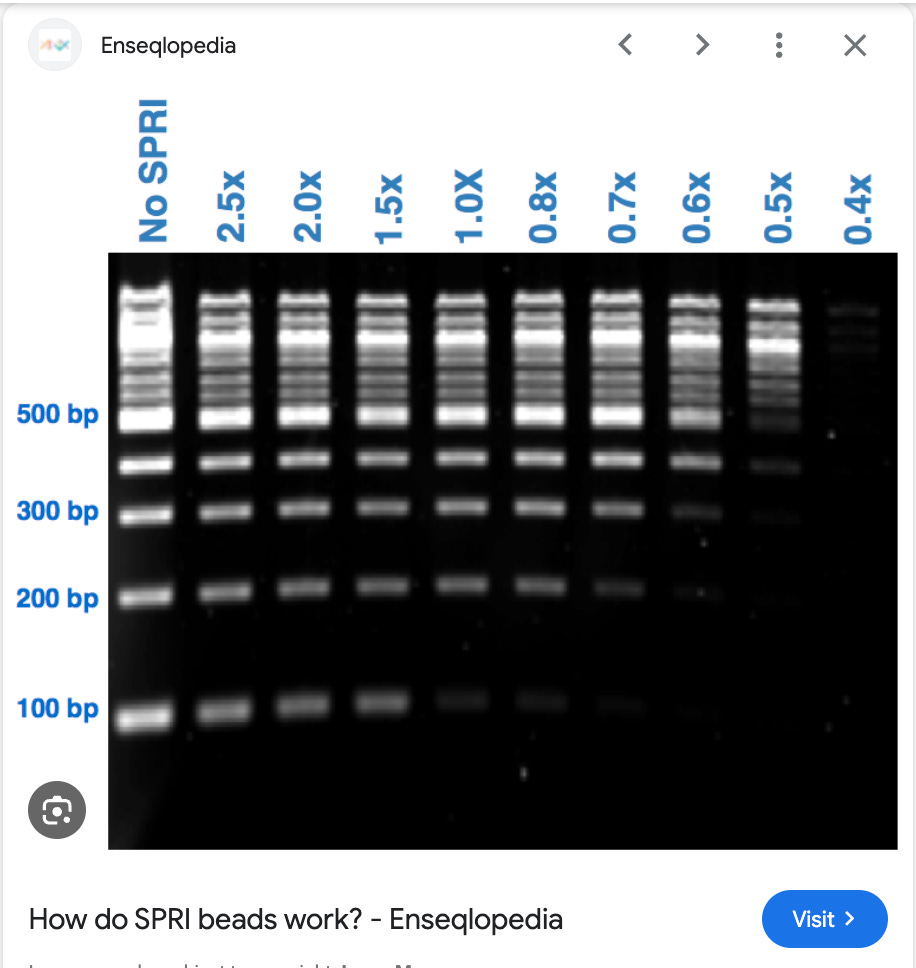
What you see above is DNA ladder being prepped with SPRI (AMpure). SPRI stands for Solid Phase Reversible Immobilization which was first published by Trevor Hawkins (DeAngelis et al) who was an early mentor of mine on the HGP. After Trevor left the HGP, we carried his torch and extended using these beads to prep plasmids.
This was trickier as we didn’t want the 6Mb E.coli genomic DNA, we didn’t want the under 500bp shrapnel but we did want to isolate DNA right in the middle size range of 5-10kb.
It required a 2 step process where we bound chromosomal DNA (6Mb E.coli DNA) with mild precipitating conditions to one set of beads and then processed the supernatent of that bead capture (smaller stuff that didnt bind to the 1st beads) with another set of beads under more stringent conditions that could bing >2Kb.
We then built this factory floor to repeat it 10 million times on 10 million plasmid, each with a piece of the human genome cloned into them.

These were minimal plasmids. They only had an antibiotic resistance gene and a bacterial origin of replication and all types of sequences to prevent the expression of the human DNA in an E.coli cell.
We wanted Zero Promoters as human peptides expressed in E.coli could be lethal and that would create a gap in the human genome reference sequence.
The reason I'm belaboring this point is that some of the people trying to quantitate the plasmid DNA in these shots, don’t seem to be aware of the limitations in the prep kits they are working with.
As a result they are loosing a large portion of the DNA in the DNA prep itself. It's a clever trick that will probably slip through Peer Review.
Take this image as an example.
This is NEBs plasmid prep kit.
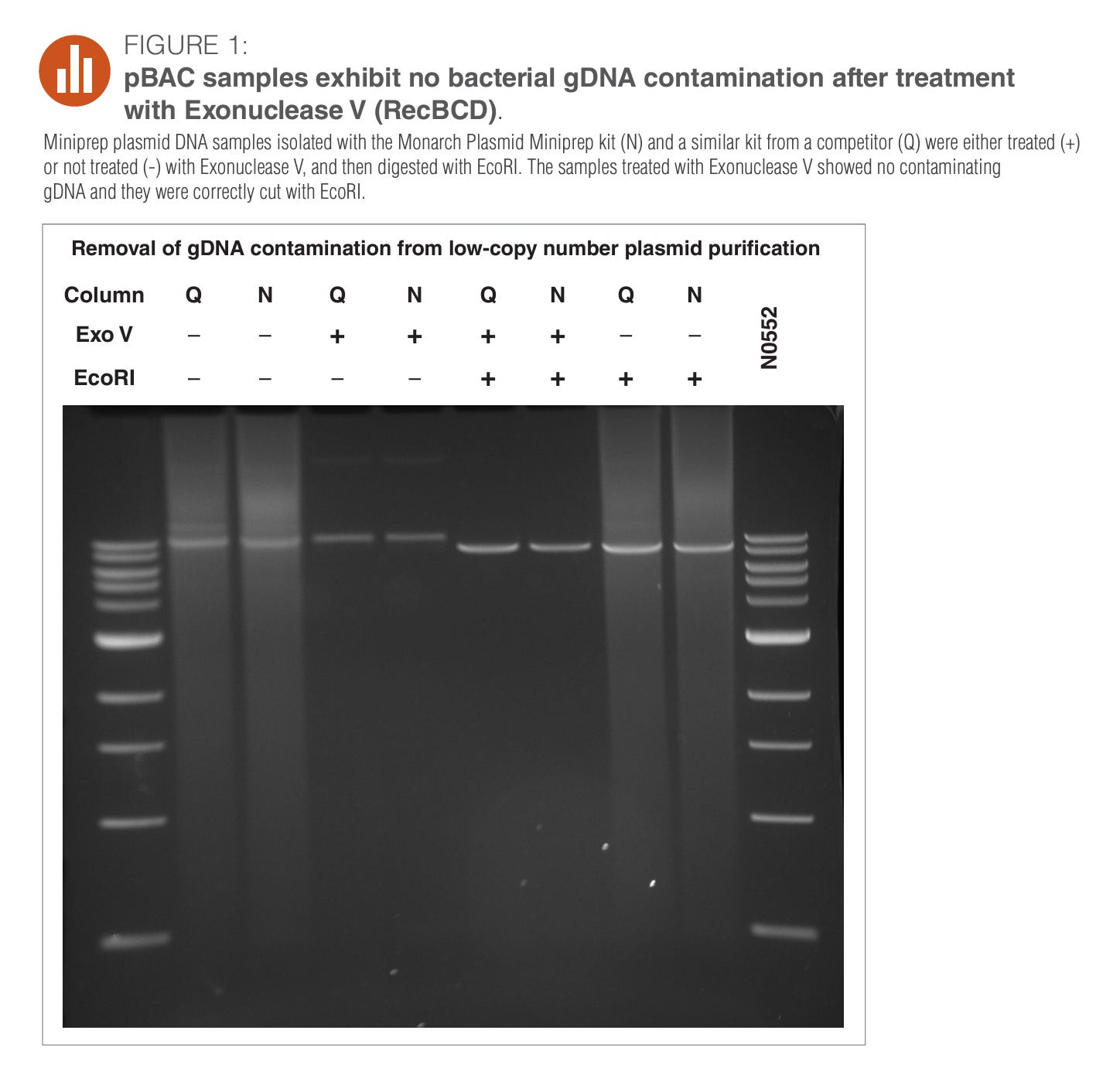
The Q and the N notation on the Column likely stands for Qiagen and NEB.
When those are negative (no prep) you see a smear that extends to the lowest (smallest in terms of base pairs) band on the gel. Once you prep it (+ columns in Q and N), all that shrapnel is gone. So if the vaccines are DNaseI’d and turned into a shrapnel smear, you loose the smear in your attempts to free the DNA from the LNPs with your DNA prep.
Great way to claim your vaccines don’t have any residual DNA!
This has already manifested itself in the Kaiser et al paper where they use an EtOH precipitation without any controls to monitor the loss of the yield of the small fragments in their DNA prep. What that study needs is a 10bp DNA ladder control that is put through their DNA isolation procedure to prove they are actually capturing the small material with their prep.
Or they could just heat the sample or apply Triton-X and none of these Dirty Deeds would nullify the results.
Keep an eye on this. With so many papers emerging showing there is a problem, I expect Pharma and regulatory agencies to fund studies that will use this trick to claim there is nothing to see here.

Kevin, great picture of 320 Charles St. production floor! I have been working there for the Human (and mouse) genome sequencing project. I also designed tiny small vector (less than 1kb) to carry chunks of DNA and improve the payload ratio. Still bothers me why would they use such large vectors for the C19 vaccines with lots of extra parts they didn’t need. Were they just inefficient, incompetent or just dumb? One has to wonder
Exactly! The results of your study are only as good as the methodology you use. So often methods are not scrutinized closely enough during the peer review process.