Failure of the linearization reaction in the Pfizer bivalent vaccine manufacturing process

The EMA documentation from
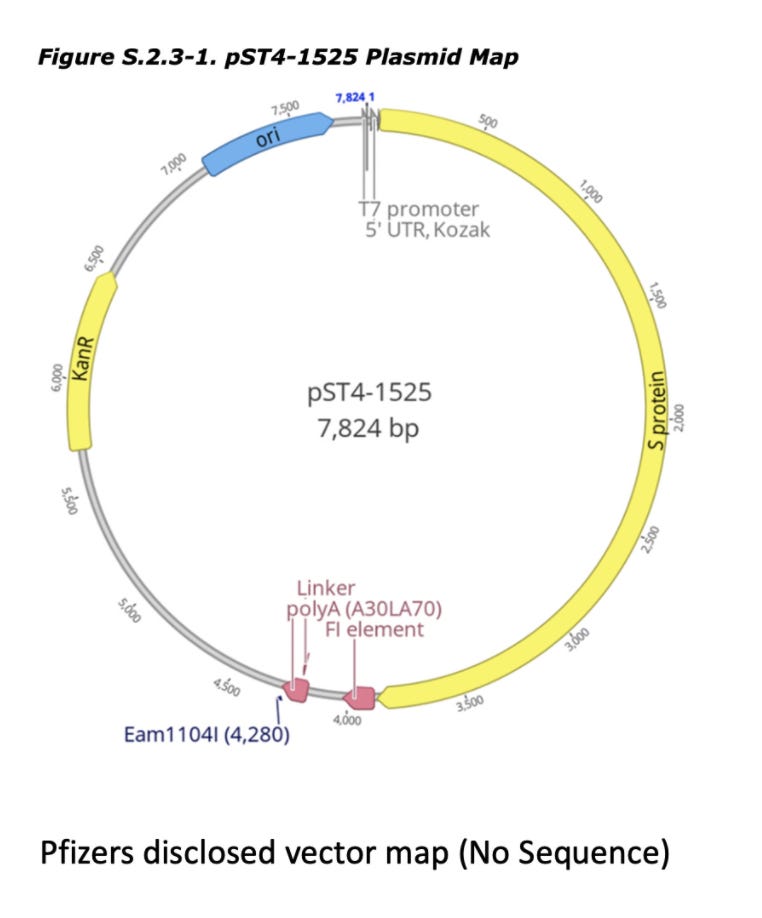
discloses Pfizers linearization strategy which utilizes a TypeIIs restriction enzyme known as Eam1104i. Restriction enzymes cut DNA based on a specific sequence pattern. Eam1104i cuts at 5’ CTCTTCN^NNN 3’ on the top strand and 3’ GAGAAGNNNN^N 5’ on the bottom strand. This can be seen in the Pfizer provided vector map at base 4,280 or ~6:00 above.
Lets looks through the Deep Sequencing in IGV and see if we can find intact molecules. Upper left is an obvious place where the enzyme Eam1104i is cutting as you can see the consistent end points in the molecules we sequence. Upper right is another group of reads where the cut sites are intact (grey = sequence that matches reference).

Let’s zoom in on that so you can see the details. Sequencing OGs will notice there is more sequencing error near these sites as we near the poly As and some sequencing adaptors (dim ATCGs in the grey reads). What does that mean?

Polymerase slippage is something that occurs with many polymerases (strand displacement polymerases exhibit less of this artifact) that stutter in long stretches of the same nucleotide like Poly As and Poly Ts. They ‘slip’ out of frame during amplification and you get an echo effect in the sequence data. PCR exacerbates the problem. Sanger sequencing of slippage is seen in figure

There are 2 PCR steps in the sequencing methods.
1)To amplify the Library after Illumina Adaptors are ligated on
2)During Bridge PCR on the Illumina array.
1- creates inter read disagreements
2- creates intra read disagreements

1- Can create high quality reads which disagree with each other on the length of the poly A and the neighboring sequence
2- Creates sequencing clusters that have discordant DNA and not fluorescently pure. This is spectral noise on the cluster being sequenced as all molecule in the cluster dont agree.. Low quality read locations.

Since a sequence of 110As is like a jigsaw puzzle thats all one color, you want some edges to these sequences that help to define it as an edge piece or a provide some signature.
You want reads that anchor outside of the poly A on both sides of the 110bp poly A region. So you have some signature to map reads. Since we have a very short library (250bp) we can’t expect many reads to bridge 100bp poly A because ~150bp of the 250bp is Illumina adaptors.

This is why we are being very cautious on calculating the % circular DNA. If you have very short insert libraries, you won’t sample many reads that span a 110bp un-mappable polyA region. You need 100bp+110bp+100bp so you have unique 100mers on each side of the polyA. 460bp library would be preferred but we might be able to live with 360bp (50bp anchor +110bp polyA +50bp anchor+ 150bp Illumina adaptors).

We got very lucky we captured any junctions at all and for that reason any estimation of circularity will be an underestimate. You'll notice out average insert size was 105bp.

250bp library - 150bp adaptors = 100bp inserts.

As you can see, while we learned a lot from these RNase A libraries, we now know there is a long Poly A in the vector and to properly span it we need larger insert libraries. This is as easy as making the 3minute fragmentation step down to 1minute and we should get larger inserts for our next Illumina run. This should provide a less biased assessment of the circle vs linear assessment.
Since the linearization step uses a single cut site, they could very easily quantitate the efficiency of this step with a qPCR assay that spans the cut site. The cut site is very close to the poly A tract which might make this a bit tricky to design.
They should also better disclose what assay they are using to detect residual DNA from the DNase I step. The next post will touch on this step of the process and what we can learn by using DNases and RNases in qPCR.
This pucing tour is depressing but, nonetheless I pfind it pfascinating. Thank you for all you are doing. Miss being able to see soothing kitty pics on my feed to help me cope. When the world is melting down, there's nothing like a cat to reassure you life is still purrty great.