

Viral and Viroid replication and the impact on genomic surveillance.
What can be learned from sgRNA and gRNA ratios in a cell?


Some questions have emerged lately regarding the fidelity of viral replication and the impact on quasi-species swarms. To summarize, the diversity of the swarms is directly related to the error rate of polymerase. The error rate of the polymerase is a governor for genome length with larger genomes requiring higher fidelity polymerases and smaller ones allowing for lower fidelity polymerases. Higher error rate = increased swarm diversity and smaller genomes with the 254bp viroids being the most diverse. Error correcting polymerases like the ExoN component of RdRp in SARs-CoV2 RNA are some of the longest RNA viruses and have less diverse swarms. DNA viruses have even higher fidelity than RNA viruses and can maintain larger genomes like the 1.26Mb Megavirus chilensis.

Viral genomes may contain several genes which the virus may need to express at different magnitudes. As a result it doesn’t express the full length genome into order to do this. It specifically expresses each gene discretely my making subgenomic RNAs (sgRNA).
Think of it like indexing a book with chapters. You may need 400,000 copies of Chaper N, 300,000 of Chapter E, and 200,000 of chapter Spike and only 2,000 copies of the chapter called ORF1ab. These shorter sgRNAs have a 75bp leader sequence attached to the 5’ end that is derived from the 5’ end of the viral genome. Think of this as “Express Me” sign attached to the beginning of each gene. Kim et al has a good color coded depiction of the Leader sequences in Black being ligated to the front of each gene.
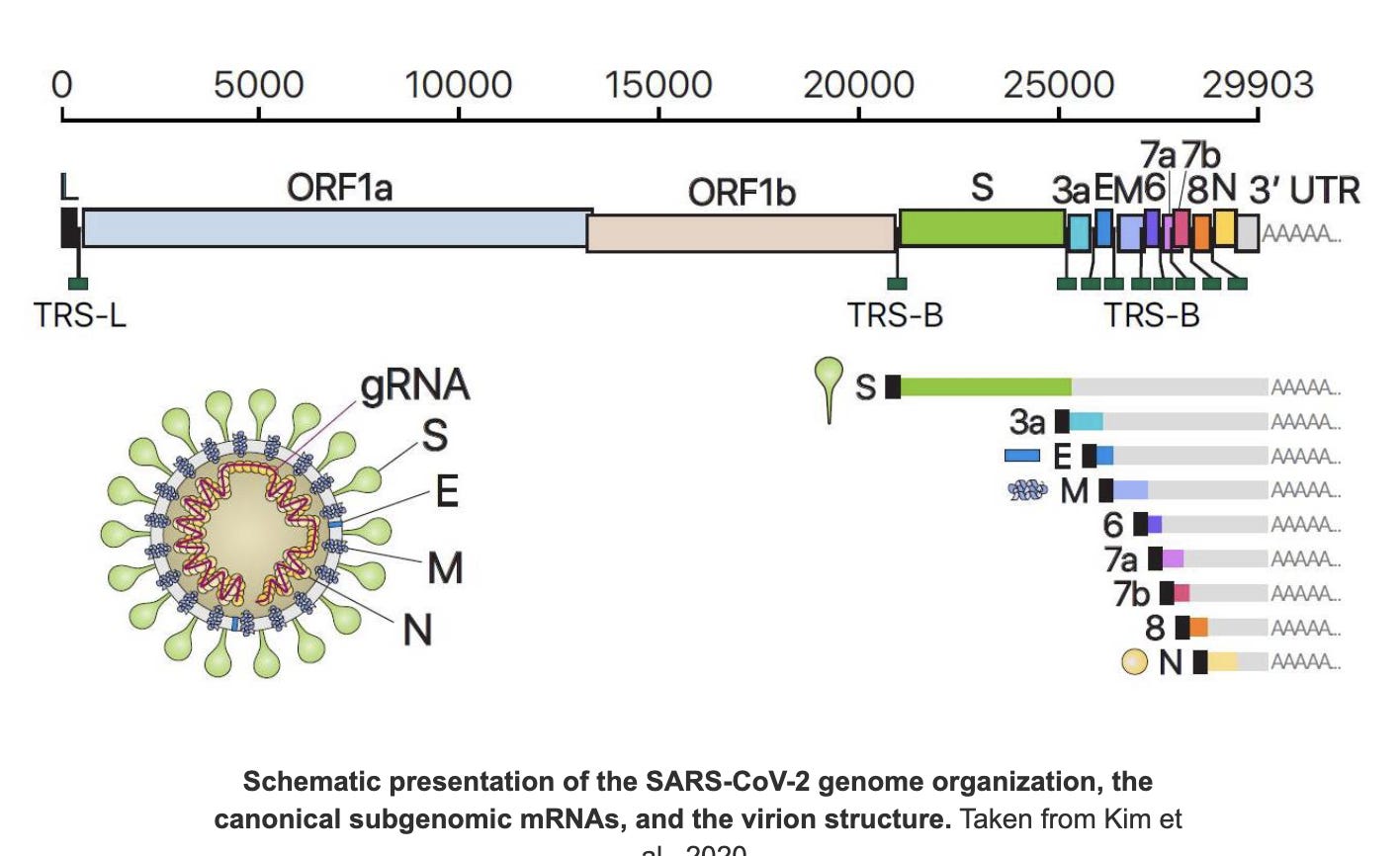
However, each virus also needs to make at least 2 copies of its entire genome for it grow exponentially.
In case of C19, the N genes is the most highly expressed which explains why it the most targeted region of the C19 genome for qPCR assays. If you want the highest sensitivity test, you target the most highly expressed region of the genome with an RT-qPCR test.
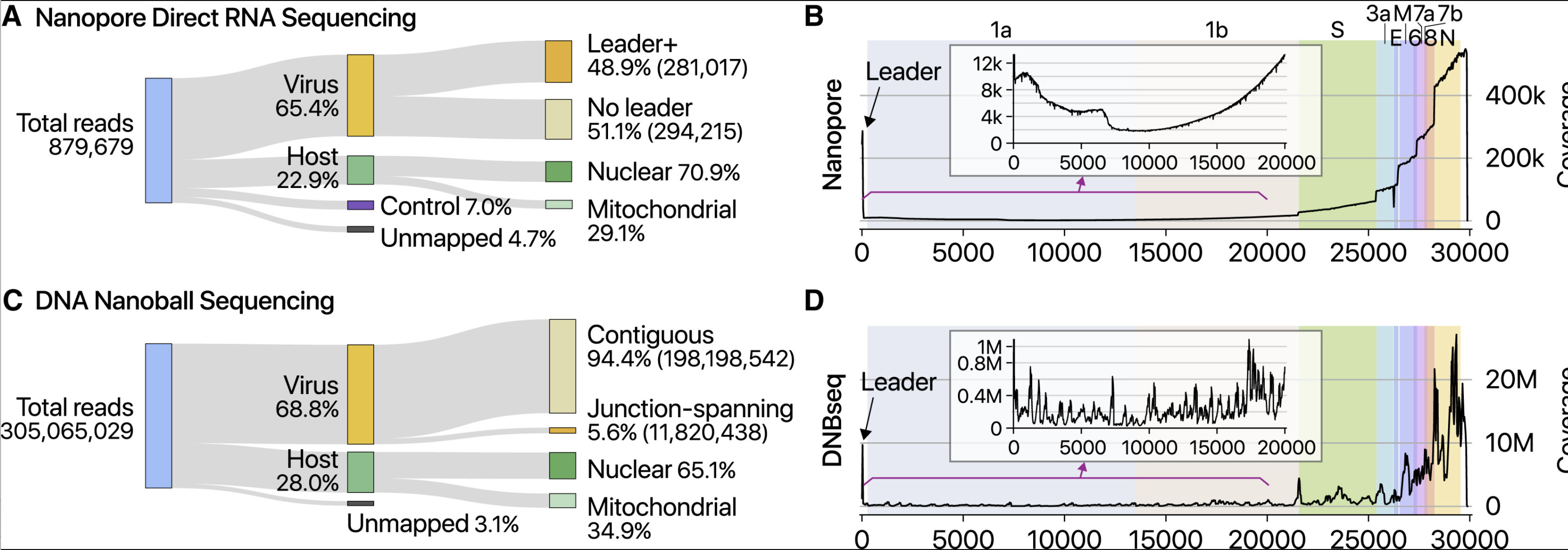
In order for the viral genome to replicate and package into more virions, it will need to make a few full length copies of its own genome in addition to coordinating this symphony of differential subgenomic RNA expression.
So what are the ratios of the the full length genomes (gRNA) compared to the truncated sgRNA transcripts that are made to drive the N gene to such high expression levels?
Does the ratio of the gRNA to the sgRNA provide us any insight into replicative cycle and or the infectivity of the virus?
Do any of the sgRNAs get packaged into pseudovirus or satellites and do the expression of such particles participate in seropositivity or T-Cell immunity in a herd setting?
There a few papers that help address these important questions but it is important to keep in mind which RNAs are replicative (gRNA) and thus contribute to the replicative evolution and transmission of the virus and which are evolutionary dead ends (sgRNA).
Viroids and Viruses can also transmit cell to cell through plasmodesmato (Plants) or tight junctions (mammals). These are communication pores that bridge neighboring cells and PTSVd is known to down regulate Callose synthsesis and thus make the plasmodesmato more porous to enable to cell to cell forms of transmission. C19 is believed to analogously impact tight junctions. While gRNA and sgRNA may transmit cell to cell this way, virions are needed to spread to another host via aerosol transmission.
This paper from Garcia-Blanco is another perspective on the ration of gRNA vs sgRNA.

A few interesting studies Garcia-Blanco et al discuss is the different gRNA/sgRNA ratios you see depending on if you centrifuge (clarify) or don’t centrifuge the sample. If you pellet cells, and test those you get different gRNA/sgRNA ratios than if you qPCR from the supernatent of the centrifugation. Virions with gRNA should remain in the supernatent. The pelleted cells with have both sgRNA and gRNA. If the sgRNAs are packaged we should see the the same ratio of gRNA/sgRNA in both the pellet and the supernatent. They in-fact see a large difference in the cells versus the supernatent. Other papers notice many of these sgRNAs are found within DWV (Double Membrane Vesicles) with in the cells and this may lead to further nuclease resistance and persistence of these molecules. You can see how these slight differences in protocol may lead to vast differences in sequencing studies attempting to survey this problem.

While Kim et al observed 400,000X coverage over the N gene, they only observed 111 full length 29kb viral RNA sequences. It is well known that the Nanopore systems have a bias for sequencing the shorter molecules more readily than the longer ones. There is a whole industry focused on how to purify DNA so people can get megabase reads from a nanopore. You have to purify 150Kb DNA in order to average N50 readlengths of 30-57Kb. Even with gel purifications and sizing of 150Kb bands, the pores vacuum up all the small stuff first.
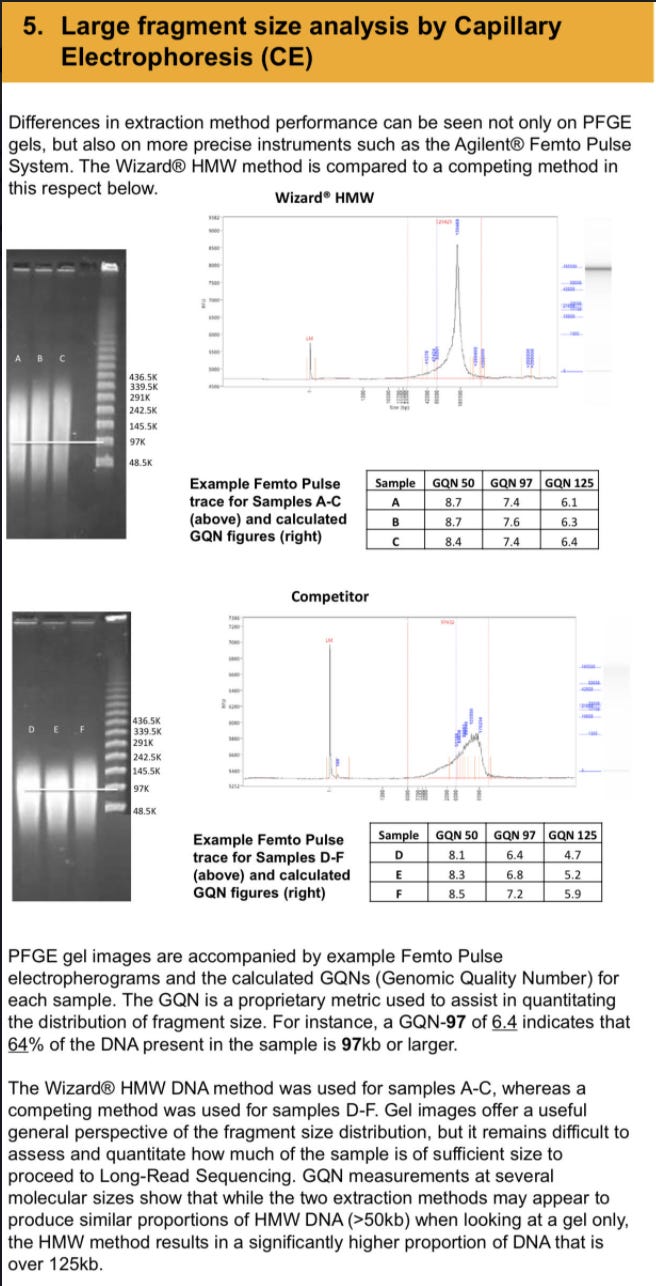
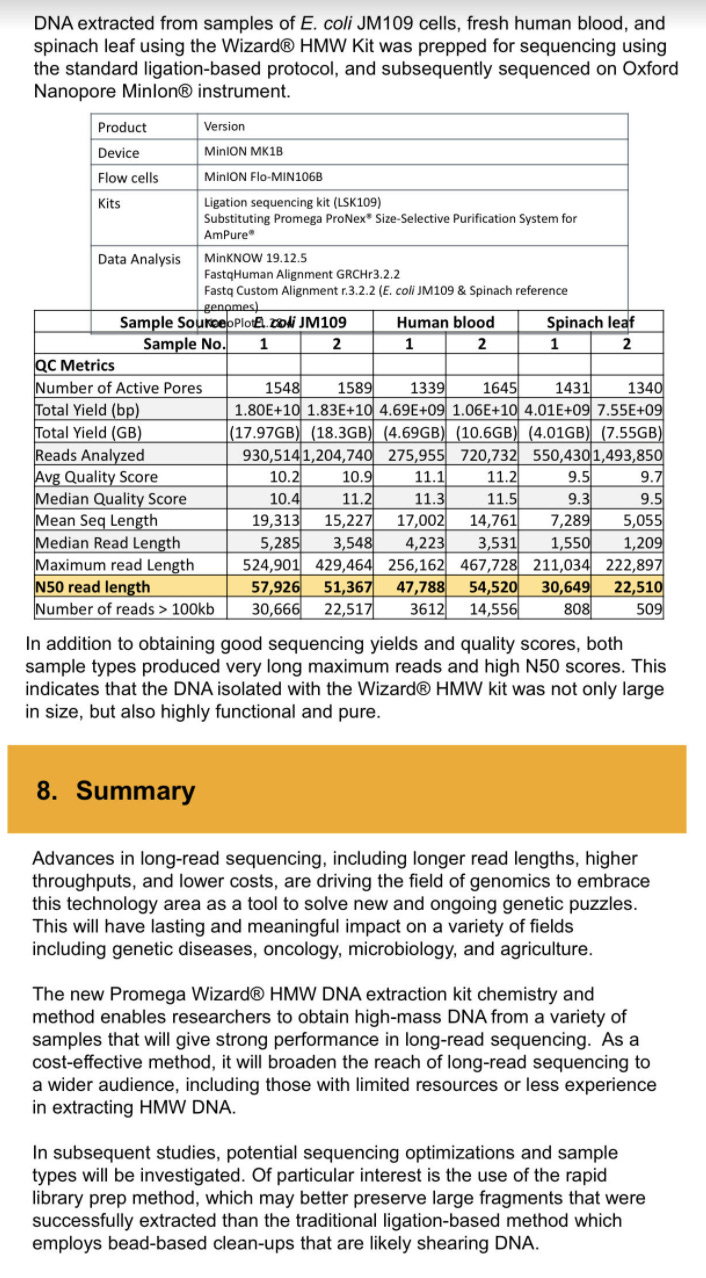
As a result of this artifact in Kim et al, digital qPCR methods are used to more finely quantitate these gRNA/sgRNA ratios.
Many of these studies are referenced in Garcia-Blanco and the consensus points to a 3-5 order of magnitude more sgRNA than gRNA in cells but mostly gRNA in supernatents.
One of the reasons I have been droning on in the twitter verse about live-dead qPCR is because the qPCR assays that simply target highly expressed genes tend to be positive for weeks after you are no longer infectious. Many years into the pandemic the CDC finally put a 90 day moratorium on qPCR testing. Once you are positive, there is no point testing again with N gene based qPCR for 90 days later or your positivity will simply be assumed to be a result of the residual sgRNA you have floating around from the prior infection.
Prior to this moratorium, this had massive consequences by inflating the size of pandemic. Recall anyone positive with sgRNA based qPCR will get flagged as a COVID death even if they fully recovered and died of something else. Any asymptomatic qPCR positive case that was fully recovered would also get flagged for isolation.
This was differently enforced in each jurisdiction but some had 28-day positivity windows. Even the CDC admitted that to nearly half of the COVID deaths being “with COVID” not from it. Other studies have shown the average age of death “from COVID” to be the same as the average age of death (80). Studies have found over 94% of C19 deaths have 2-3 other comorbidities. This mess has led many statisticians to wisely focus their efforts on excess mortality metrics.
So while many people engage in these drawn out debates regarding if the PCR tests are hitting other coronaviruses or influenza, this is a complete smoke screen of the real problem at hand. Even if you are 100% accurate with nailing SARs-CoV-2, until you start differentiating the infective virus from the corpses, you have enough false positives to create a casedemic. If you are infectious for 9 days and can be PCR positive for 90 days, 90% of the 100% accurate tests will still be quarantining people with C19 in the rear view mirror.
These are people that are immune to C19.
What happens when you quarantine 9 immune people for every infectious person qPCR picks up?
You guarantee herd immunity is never reached!
You need the immune people back in the herd ASAP for herd immunity to work.
So the debate over the late CT PCR assays hitting other coronaviruses is all squid ink. It doesn’t matter. Its poor form and a sign of haste but sharpening that knife wont get us out of the doom loop. Even if you are 100% accurate with a test that chases C19 corpses, you maximally delay herd immunity.
It is the ideal strategy if you want to maximize testing and vaccine revenue.
Coincidentally N3 primer on the CDC test back in January 2020 was binding to human DNA.
Thanks for posting this Kevin.
I recently came across this presentation by Matt Parker from the Sheffield Biomedical Research Centre in the UK titled "Altered Subgenomic RNA Abundance in SARS-CoV-2 B.1.1.7 Infections"
https://www.youtube.com/watch?v=aK8zp57FIK0
I found it pretty interesting but would welcome your thoughts if you have time.
Best wishes
Lee