
DNA-RNA hybrids, R-Loops and nuclease resistance of the mRNA vaccines
DNA-RNA hybrids, R-Loops and nuclease resistance of the mRNA vaccines


Introduction
To better quantitate the number of full-length and circular DNA molecules in the vaccines, Jonathan Gilthorpe suggested a good experiment with T5 exonuclease. We were drawn to this approach as our lab does not have an electroporator commonly used for transformation of E.coli. We can heat shock E.coli but this isn’t the most efficient means of transformation and thus will under quantitate the DNA. While we ran a quick and uncontrolled experiment with heat shocking E.coli cells with these vaccines, we did not sequence verify the colonies to confirm they are transformed with fully intact expression vector. This is an important step for confirmation of the transformation.
We are collecting reagents to perform this test more quantitatively. As supply chains challenges continue to plague our field, we took an interest in a simpler method proposed and we learned something from it.
The alternative suggested approach was to use enzymes that destroy non-circular DNA and qPCR before and after these enzyme treatments.
T5 Exonuclease will degrade any DNA that is not circular which should result in only DNA that is E.coli competent. However, T5 exonuclease will also destroy nicked plasmids which are still competent (albeit -100 fold less) and thus will also slightly under quantitate the competence of the residual vaccine DNA. Frequently, the conditions (alkaline lysis) used to lyse open E.coli, will nick the plasmid DNA but any molecule that is still circular will be resistant to T5 exonuclease.
Keep in mind this is a very E.coli centric question as Bacillus can be transformed with linear DNA.
If you perform qPCR with and without T5 Exonuclease (NEB #M0363) on the purified nucleic acids from the vaccine, you ‘can in theory’ zero in on how much fully circular, non-nicked plasmid DNA is still around.
In theory…
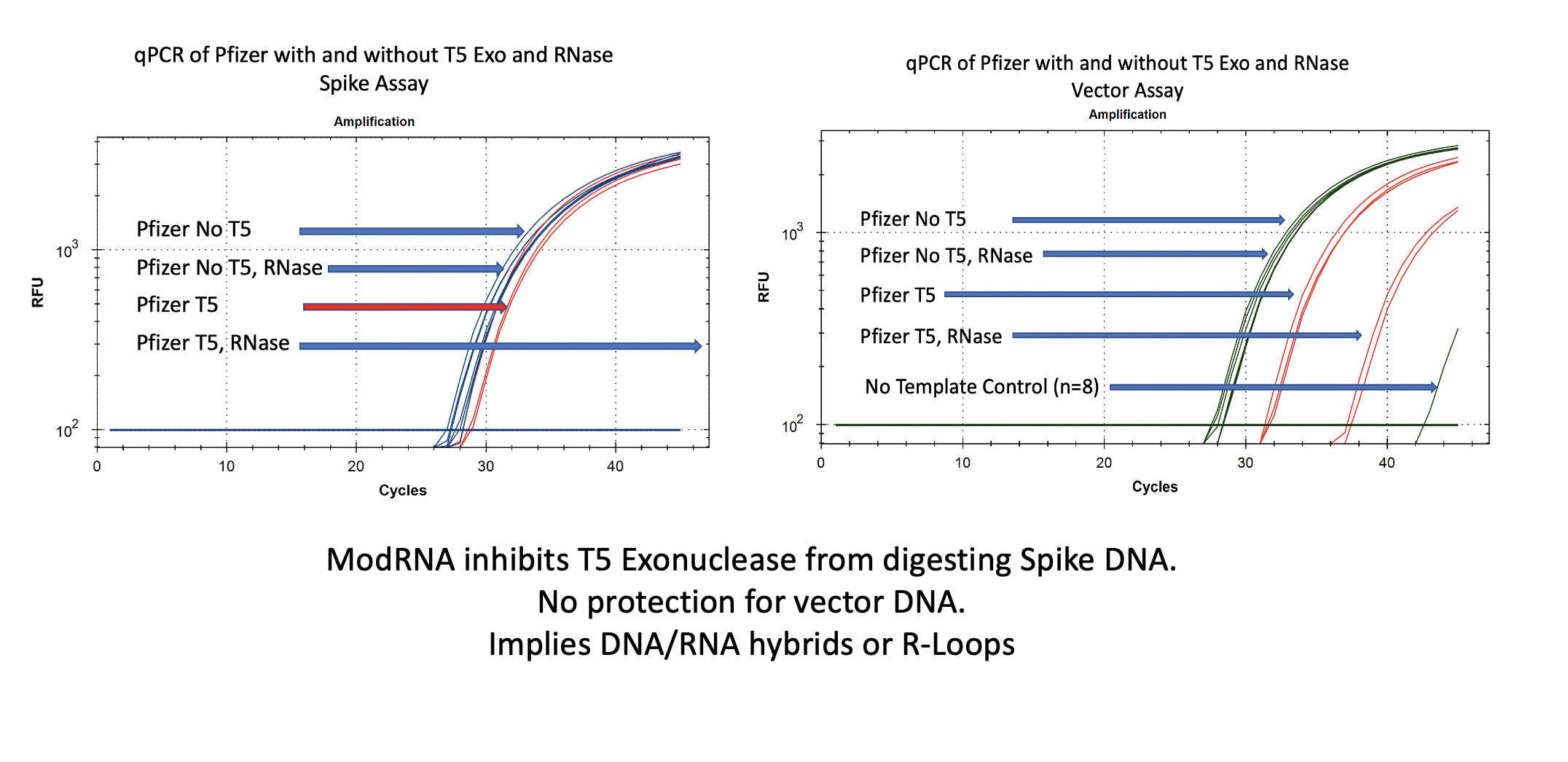
You will notice in Figure 2 that RNase A does not move the qPCR signal on the vector (right) or spike (left) DNA. This is expected as RNases do not digest DNA.
In the case of the Spike sequence, we see ~1-2CT shift on T5 exo treatment (Left red lines). Once the RNA is removed with RNase A, the spike DNA is completely eliminated.
The vector DNA is more nuclease sensitive. T5 exonuclease moves the CT from 28→31. Without additional controls and looking at both vector and spike sequences, one might think that 3 CT offsets implies 8 fold (2^3CT) more digestible DNA than circular DNA and thus lots of circular DNA!
Not so fast.
Additional controls on the vector nuclease experiment demonstrate prior removal of the RNA enables more T5 exo digestion of the DNA. The difference between the digestible DNA versus the non-digestible DNA after the modRNA is removed is much more striking. We see a 9 CT shift (28CT to 37CT) as opposed to 3 CTs. An 8 fold reduction is now a 512 fold difference once you remove the modRNA.
This is also seen with DNaseI. This is the enzyme used by Pfizer to remove the plasmid DNA according to the EMA disclosure. You can see that the DNA for the vector is easily cleared (CT over 30) with DNase I but the DNA for the Spike remains at CT of 22-24 after DNase I treatment. If you RNase A this sample to remove the modRNA prior to DNase I treatment, the samples show a 10CT offset (1000 fold reduction).
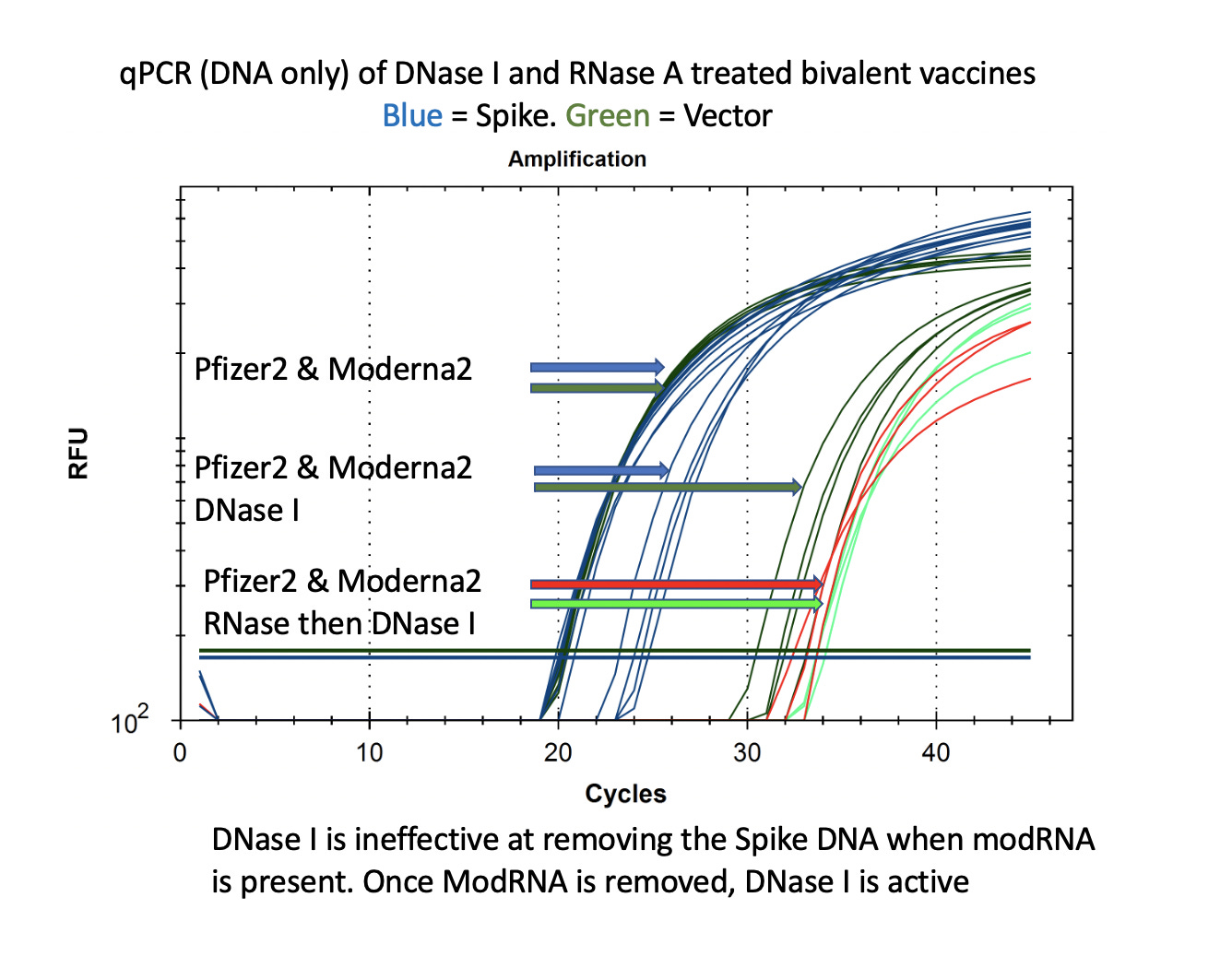
It is important to note that the Spike sequence is nuclease protected and the Vector sequence is not. This implicates the modified spike mRNA as a nuclease protectant for the DNA that it is expressed from.
Why is this happening? What protection could the RNA offer the DNA in the presence of aggressive nucleases?
This brings us into the realm of RNA/DNA hybrids and R-Loops. The RNA:DNA hybridization is favored over DNA:DNA hybridization and this especially true in the case of N1-methylpseudouridine (m1Ψ). Thus the DNA contamination is likely partially hybridized to the modified RNA and this makes it nuclease resistant.
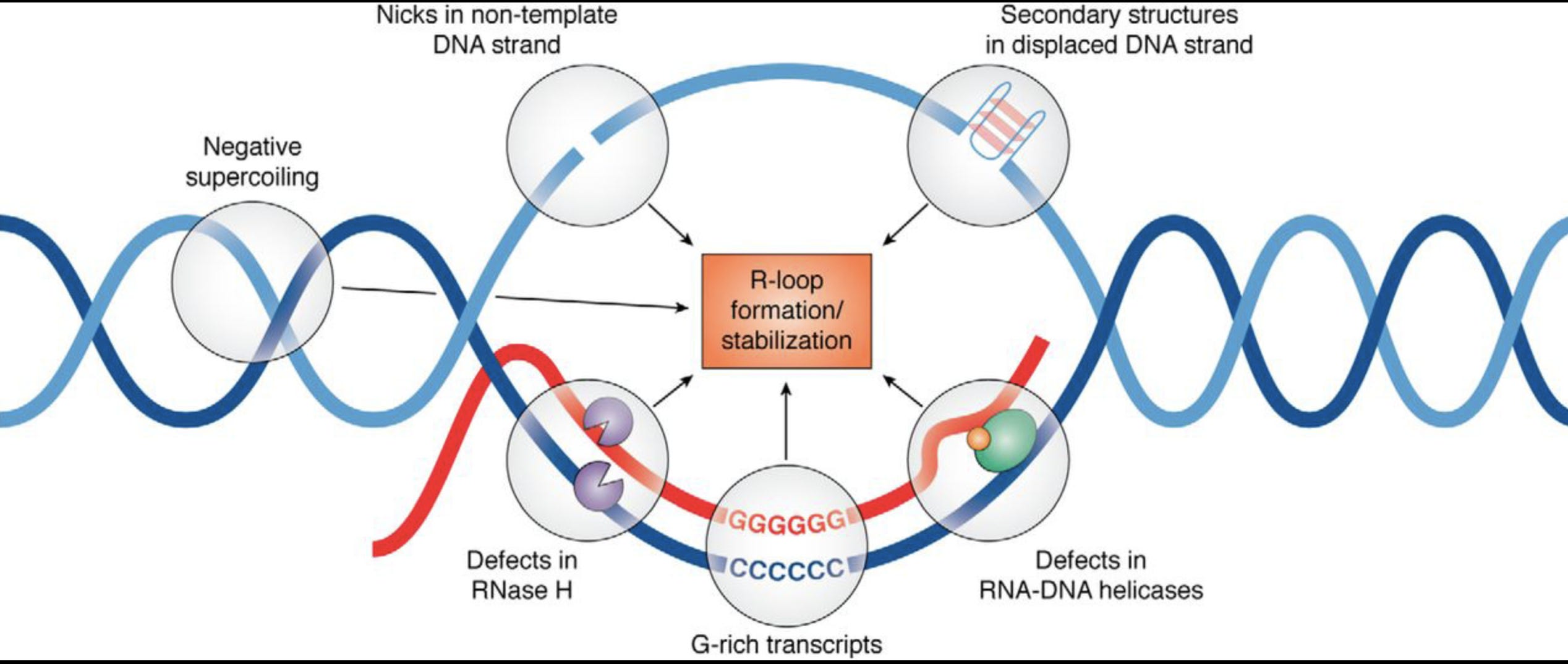
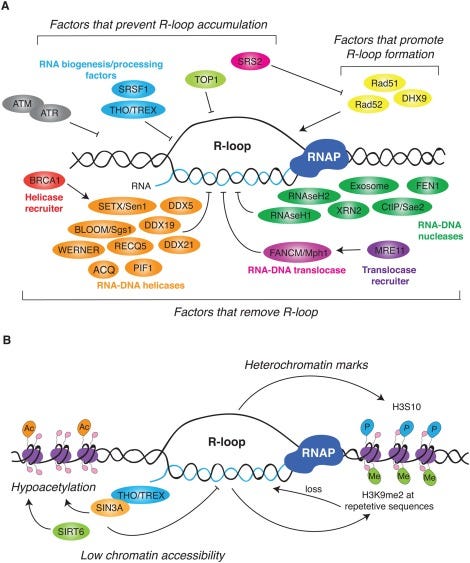
David Wiseman asked a pertinent question. “If your lab nucleases are confused by this, what happens in a mammalian cell transfected with these?” Good question. This brings us to Crossley et al. that demonstrate RNA-DNA hybrids active immune responses and trigger a host of other pathways.

Keep in mind your cells are processing R-Loop all the time. Every gene that gets expressed generates these, however these modRNAs are not ordinary genes. As we have shown, they stall nucleases, they are quadruplex G dense and the 801 m1Ψs is not something the cell has ever seen before. m1Ψ is usually at 0.2-0.6% in human mRNA. The vaccine mRNA is 100% m1Ψ.
Why does m1Ψ matter in the world of R-Loops and RNA-DNA hybrids?
There is an important paper from Callum Parr et al. on the changes in melting temperature with the introduction of m1Ψ.

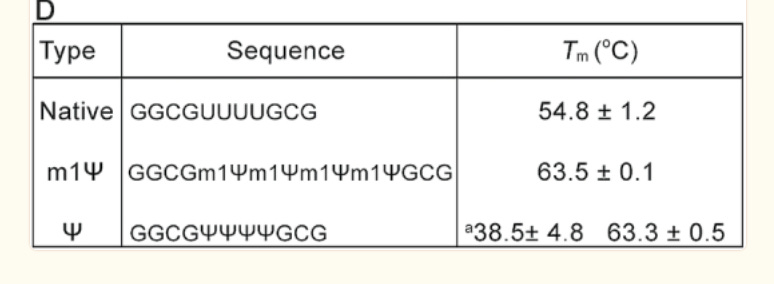
Note the 9°C change with the addition of just four m1Ψ into a sequence. The Pfizer vaccine has 801 m1Ψs. Higher melting temperatures mean the RNA-DNA hybrid is stickier and requires more heat to denature. We covered some of these issues in a prior preprint. This makes these modRNA resistant to RNase-L and very ‘sticky’ to DNA.

The codon optimization raised the GC content of the vax spike sequence, introduced quadruplex Gs and altered the secondary structure of the mRNAs. The m1Ψ exacerbates this secondary structure. These DNA/RNA hybrids may be challenging for the cells to clear. Some of the unexpected persistence of these vaccine molecules in patients could be the result of contaminating DNA/RNA hybrids that our cells have a hard time clearing. This may not have affected the vaccine trials as those trials were not run on plasmid generated templates but were manufactured with linear DNA attached to magnetic beads which may have been easier to remove.
What does m1Ψ do to R-Loop processing? It appears R-Loop processing is in the BRCA1 pathway and is integral to genome stability, cancer, and neurodegeneration.

Conclusions
T5 Exonuclease and DNase I (DNA nucleases) are substantially inhibited by the presence of complimentary vaccine mRNA.
The literature on m1Ψ’s impact on Tm (melting temp) implies m1Ψ is a very ‘sticky’ base and likely forms strongly bound DNA-RNA hybrids and R-Loops that are nuclease resistant. This may explains why the plasmid DNA has been difficult to remove from the vaccines. This also brings into question what SYBR dyes do in the presence of DNA/RNA hybrids and if we’re in new territory trying to quantitate the DNA content in these vaccines. We mentioned this in our Preprint as a potential limitation to the Fluorometer based measurements.
This raises questions regarding how mammalian cells sense DNA/RNA hybrids and if these hybrids are the reason this m1Ψ modRNA can be detected 28 days later in plasma.
Methods
RNase A
10ul RNase A (20mg/ml) was added to 50ul of CTAB purified vaccine mRNA. RNase A (NEB) treatments were performed at 37°C for 30 minutes.
DNase I
3ul DNase I (Grim Reefer enzyme MGC #420145), 6ul 10 X buffer, 1ul ddH20 was added to 50ul of CTAB purified vaccine mRNA. Treatments were performed at 37°C for 30 minutes. 3ul of MGC lysis buffer (MGC #420001) was added to arrest the DNase I. 50ul reaction was purified using 50ul of 100% Isopropanol, 113ul SenSATIVAx (MGC #420001), 12.5ul of 25mM MgCl2. Samples were tip mixed 10 times and magnetically separated. Two 200ul 80% Ethanol washes were performed. Samples were briefly dried at RT and eluted in 50ul ddH20.
T5 exonuclease
2ul DNA were treated with 1ul T5 exonuclease (10Units/ul), 5ul NEB buffer #4 and 42ul of ddH20 at 37°C for 30 minutes. 50ul reaction was purified using 50ul of 100% Isopropanol, 113ul SenSATIVAx (MGC #420001), 12.5ul of 25mM MgCl2. Samples were tip mixed 10 times and magnetically separated. Two 200ul 80% Ethanol washes were performed. Samples were briefly dried at RT and eluted in 50ul ddH20.
qPCR was performed as described previously.
The exhaustive replacement of U with N-1-methyl-psuedo-U and excessive GC optimisation was done with the singular myopic desire to hoodwink our cell biology and max out protein translation. Concerns raised by McKernan, Kyriakopoulos and McCullough in their pre-print https://osf.io/bcsa6/ highlighted a host of problems with this approach. Sixteen months later and backed by solid experimental science, we find out the s**t show just got a whole lot worse. The lack of foresight, consequence of action and hubris by Pfizer and Moderna is unbelievable.
"maine-coon-clease"?