

Vaccine targeted qPCR of Cancer Cell Lines treated with BNT162b2
Putative integration events


Ulrike Kaemmerer has treated MCF7 and OvCar3 cancer cell lines with various vaccines. Once transfected they performed cell passaging on these transfected cell lines to dilute out the residual vaccine and identify cells which were transfected. They performed Immunohistochemistry (IHC) on these cells and documented spike expression levels.
They then reached out to us to see if we could find any DNA or RNA in the cell lines. This thread will document what we found from a DNA and RNA level.

Staining protocol given in (Mörz M. A Case Report: Multifocal Necrotizing Encephalitis and Myocarditis after BNT162b2 mRNA Vaccination against COVID-19. Vaccines (Basel). 2022 Oct 1;10(10):1651. doi: 10.3390/vaccines10101651. PMID: 36298516; PMCID: PMC9611676.)
Below is a description of the qPCR and sequencing performed.

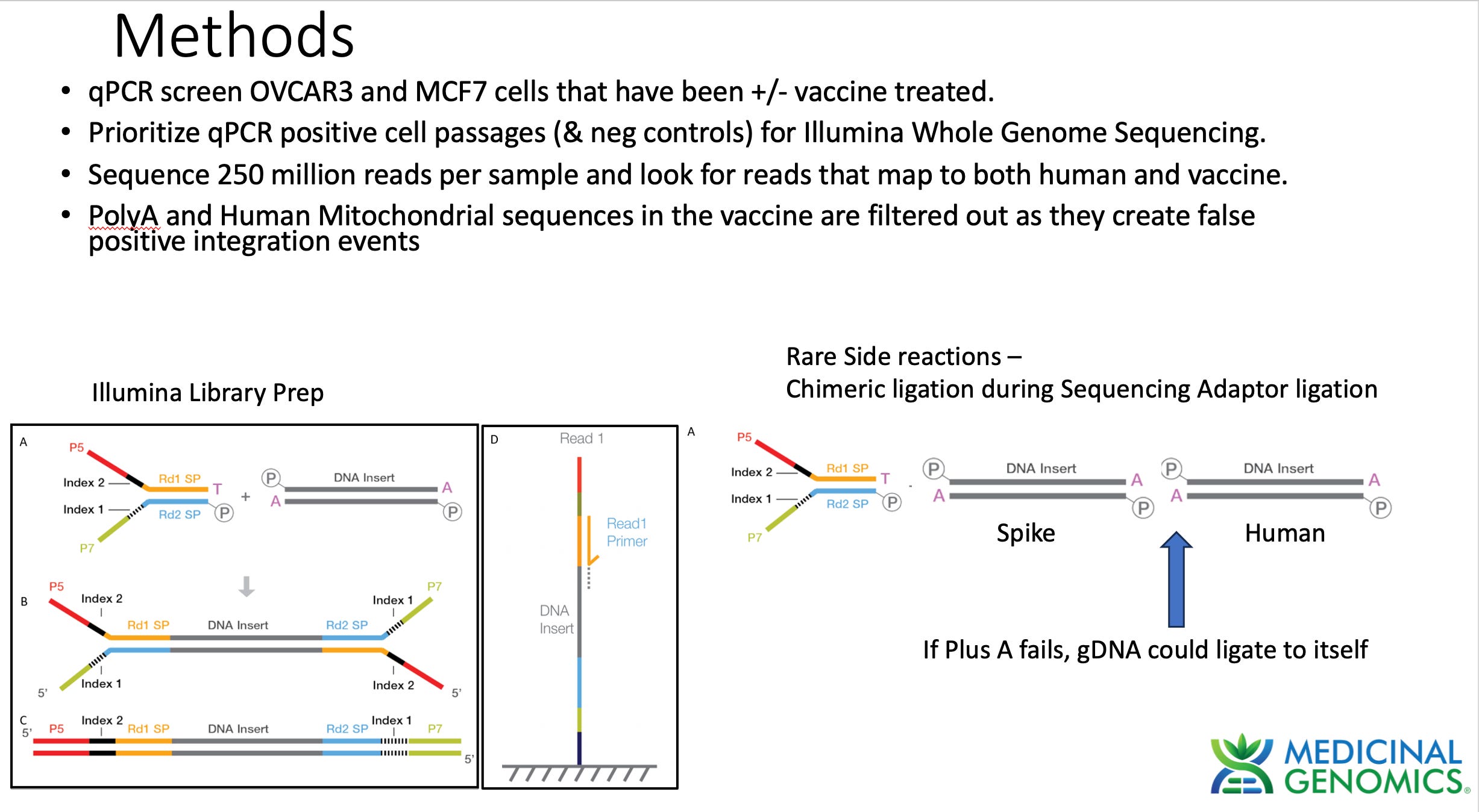
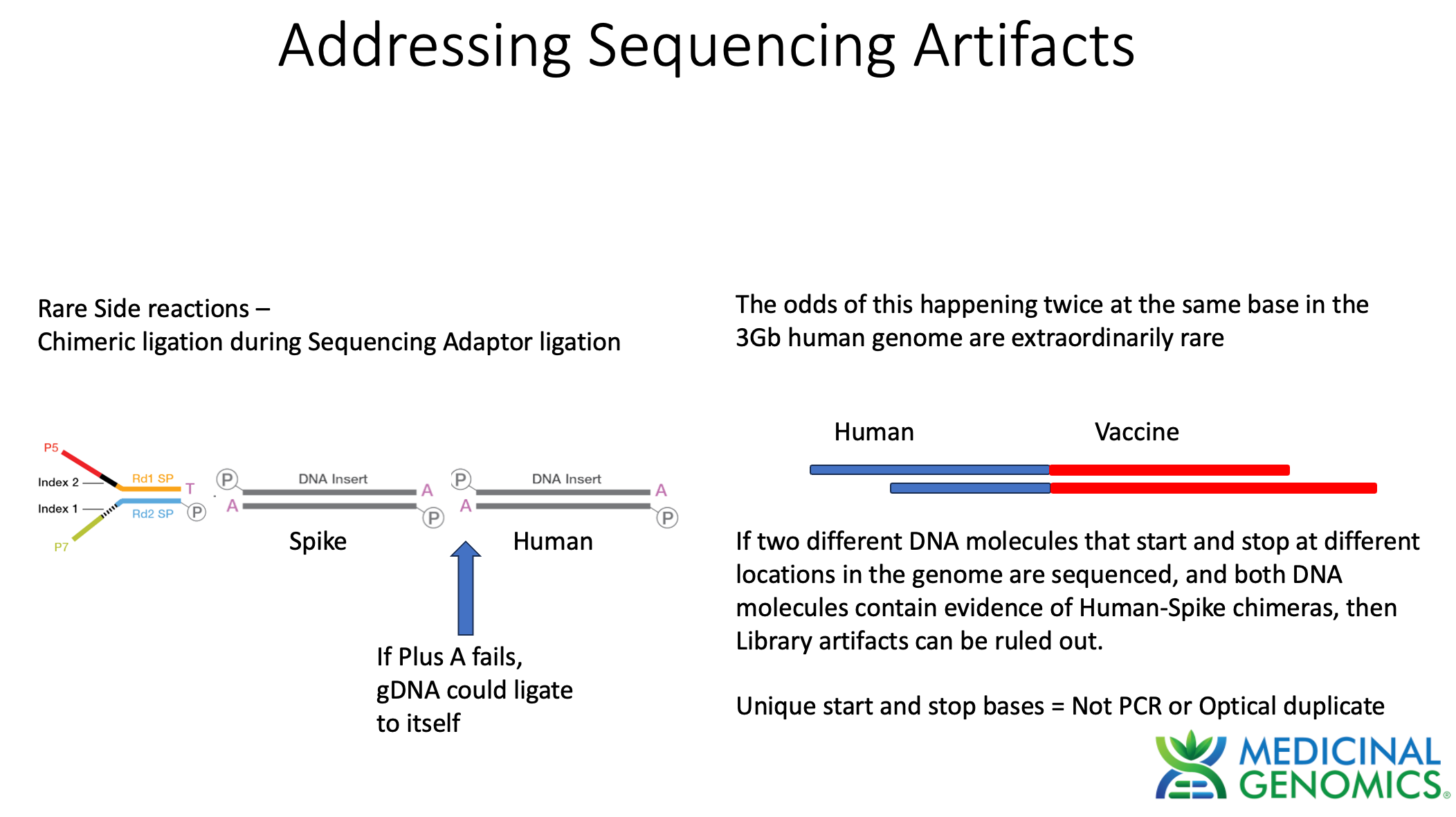
When assessing genome integration events one has to consider the potential rare side reactions that occur with Illumina sequencing. Illumina makes use of an End Repair step that adds an A to each 3’ end of dsDNA to create a Adenosine overhang DNA. This is known as a "“Plus A” reaction usually performed with a Klenow polymerase that tends to add As to 3 hydroxyl ends of blunt dsDNA.
This step is critical to ensure gDNA doesn’t ligate to itself and make chimeric DNA during ligation reactions. The sequencing primers are then added in excess. These primers have a Plus T on them. This ensures that only sequencing primer can ligate to gDNA and genomic DNA will be incompatible with self ligation. If for some reason the Plus A reaction doesn’t go to completion and some gDNA fails to Plus A, chimeric gDNA can ligate. This chimera formation side reaction should be random thus its highly unlikely for 2 fragments to ligate to each other at the exact same base in the genome. The odds of getting 2 fragments to form chimeric molecules at the same base in the genome is a 1 in 3 billion chance.
The way to combat this is to require at least 2 different unique molecules to call an integration event.
What is a ‘unique’ DNA molecule?
Each DNA fragment has a defined start and stop base. DNA molecules that have the same start and stop base can occur naturally in the DNase step used to break up the DNA into small pieces for Illumina sequencing. These duplicate sequences are often discounted as they could also arise with the PCR process used to amplify the Illumina library. These are called PCR duplicates or ‘dupes’. Tools like Picard look for reads that share the same start and stop base and flag them as potential PCR duplicates. The Illumina sequencers can also make duplicate sequences with what are known as optical duplicates. This is a software artifact that calls a single colony on the sequencing array as two colonies. As a result these sequencing reads also have the same start and stop base. They are easier to identify as their coordinates on the array are in close proximity.
There are techniques developed in the field to address this using UMIs (Unique molecular identifiers). UMIs are synthetic DNA barcode sequences you ligate on the ends of your DNA before any amplification is performed. Since every DNA molecule gets a unique barcoded added to it, you can discern if reads that share the same start and stop base also have the same DNA barcode. If they do then you know PCR over amplified that molecule. You can then use these UMIs to normalize the differential PCR that may have occurred when amplifying a library. Some regions may amplify 1,000 X and others 10,000X and this will be easy to sort out by only collecting the same amount of sequence from each UMI barcode. In our experiment we did not use UMIs so we must treat all reads with the same start and stop point as non-unique reads.
For these reasons, we do not take any singleton read as evidence for an integration event. We want to see at least 2 unique molecules with different start and stop bases covering a chimeric vaccine sequence to human sequence event to consider this a putative integration event.
Since sequencing can be costly, we first screen which cells are worth sequencing by qPCR screening the cells after vaccine treatment. Only qPCR positive cells will be graduated to sequencing.
We first start with qPCR of the vaccines before they are applied to cells to understand the quantity of the DNA contamination.
qPCR of the some of these mRNA vaccines record CT scores as low as CT 12.72!
qPCR methods were described previously.

1:10 dilutions are performed as in some vaccine vials we have noticed that direct qPCR of the vaccine itself inhibits qPCR.
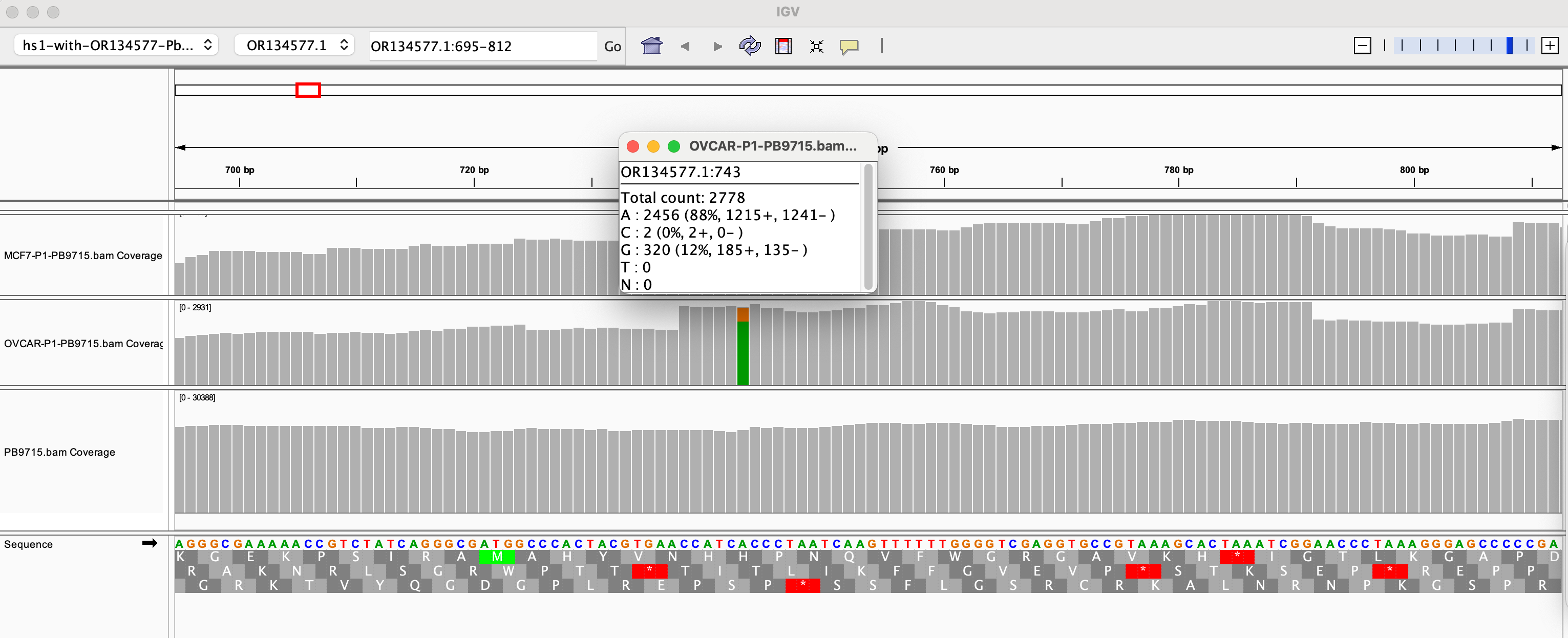
Table 2 depicts 1:10 dilutions in qPCR of cell lines treated with various vaccines. OVCAR is OvCar3. P1 and P2 represent the cell passage that has occurred. P1 = 1st passage, and P2 = 2nd passage. Mod = Moderna Lot (133 vs 140A). PB = Pfizer/Biontech with lots (9715 and 1042A).
RNAP is an internal control that amplifies a human genome. This primer set is off the shelf from IDT as it is used in C19 qPCR.
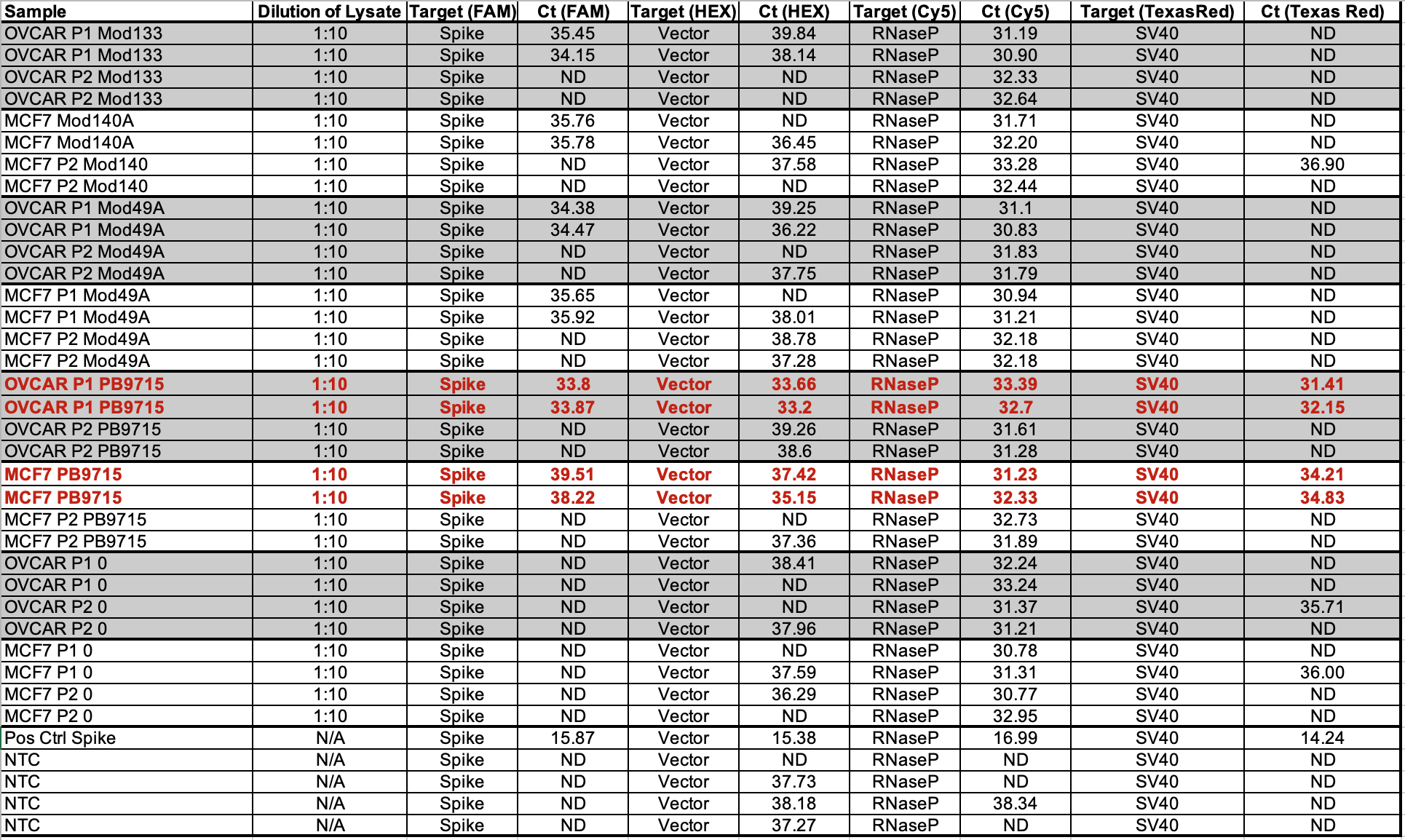
Note Moderna is not expected to amplify with SV40. The NTCs are important to pay attention to. A few NTCs are lighting up in the 37-38 range. This tells you that you shouldn’t trust any data from those channels past that CT score. The bacterial Origin of replication is known to always amplify the NTCs at CT 37+ because the polymerase used in these qPCR kits was expressed from a bacterial plasmid that has this Ori and small amounts of this residual DNA can be detected out at CT 37. There is one SV40 positive in Moderna at 36.90 while the NTCs are all clean for SV40. This could a result of SV40 having a low level contamination in those cell lines. Note this is 3-5 CTs later than where Pfizer SV40 is detected so it may just be noise.
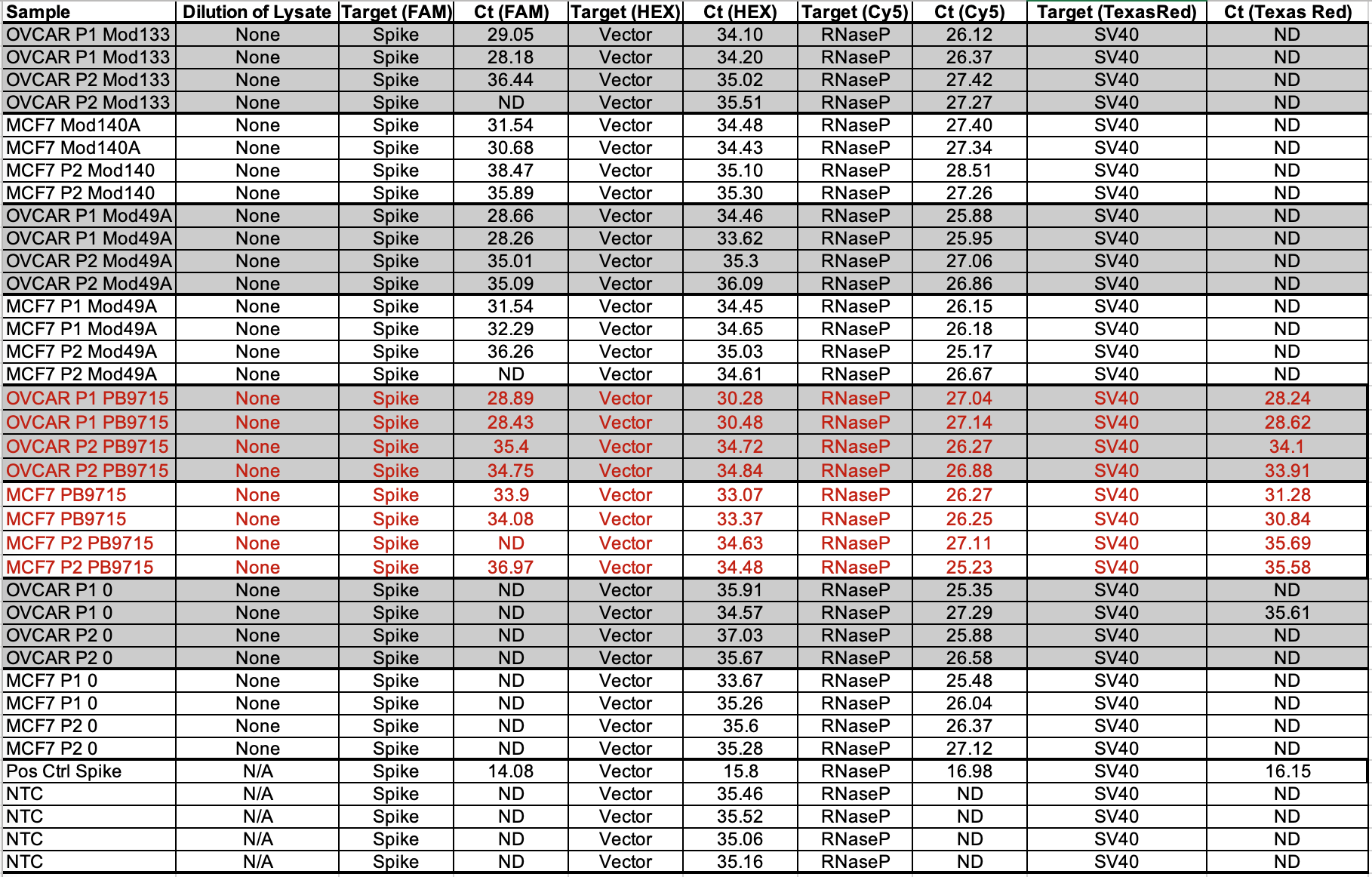
RT-qPCR of the vaccine lots are the combination of RNA and DNA signals and present even lower CT scores for SV40 (Table 3). This was RT-qPCR of the vaccines directly BEFORE they were applied to the cell lines listed.

Table 4 is RT-qPCR of the cells after the vaccine was applied to each cell line and passaged. You’ll note Passage 1 is ~5 CTs earlier than Passage 2. We don’t expect RNAP to differ between passages. Notice the vector amplicon is CT 30.28 on the 1st passage for OVCARP1 and 34.72 for the 2nd passage. ~4CTs is 16 fold difference.
Since several 100 fold dilutions (10ml washes with PBS) were performed between passages, if this were simple dilution between passages we would expect 100 -10,000 fold reduction in CT scores which is 6-12 CTs. We don’t see this which demonstrates the DNA/RNA is in the cells and not diluting out with each wash.
The qPCR data shows a larger delta CT between passages (5-6CTs or 32-64 fold dilution).
Rerun at 1:10 Dilution in RT-qPCR

Watchmaker DNA libraries were constructed according to the manufacturers protocol for Passage 1 and Passage 2 qPCR positive OVCAR cell lines. Cell pellets were lysed with 100ul of MaGiC Lysis (Medicinal Genomics part number P/N 420180) (95C for 10 minutes and 4C for 5 minutes). 100ul of stabilization buffer was added to the sample. 50ul of this 200ul lysate was purified with 150ul SenSATIVAx. Beads were separated and washed 2X with 70% EtOH. Beads were eluted in 25ul of ddH20 and 20ul used for Watchmaker library construction. Sequencing libraries were DNA barcoded and sent out to a blinded Illumina sequence provider








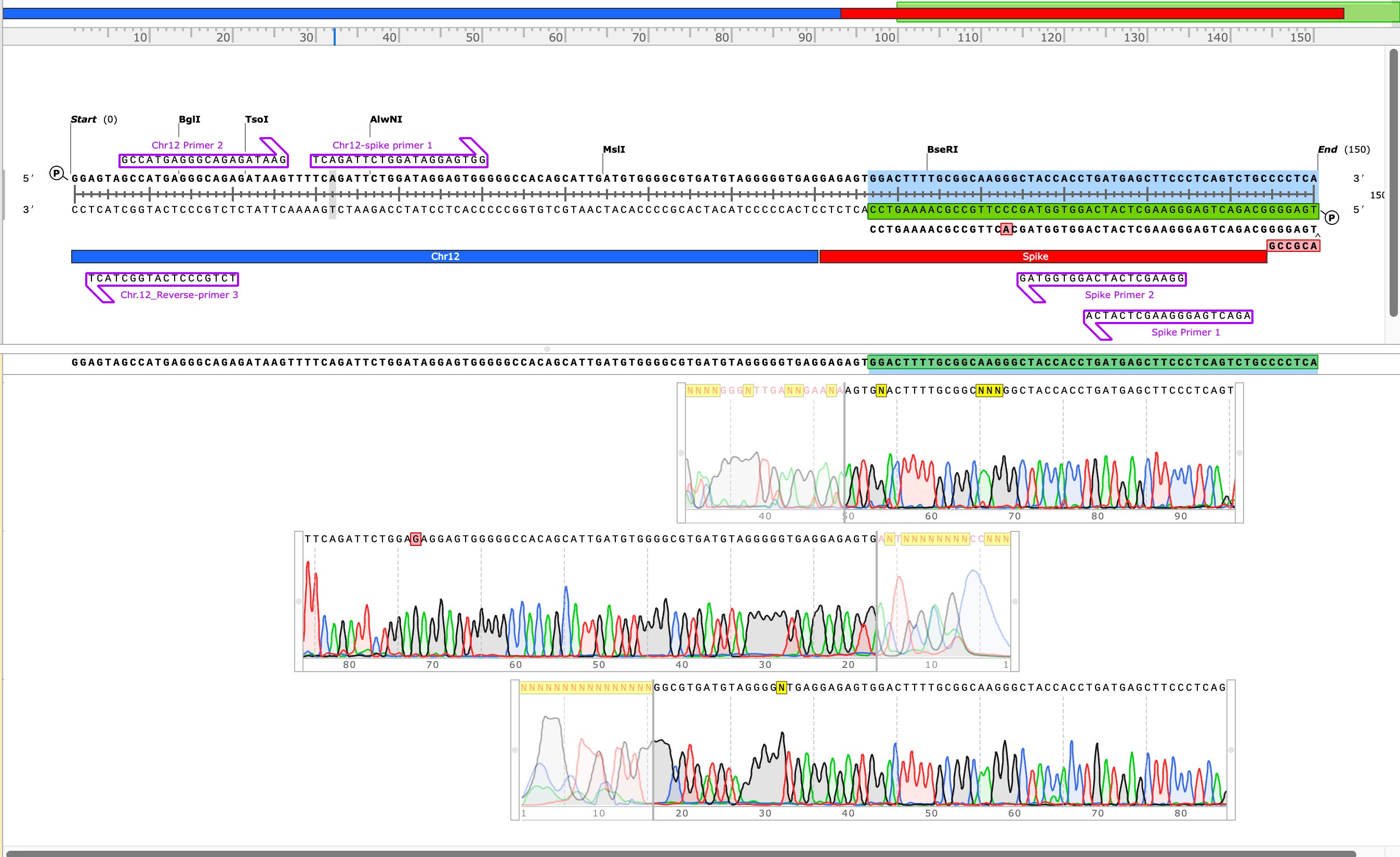
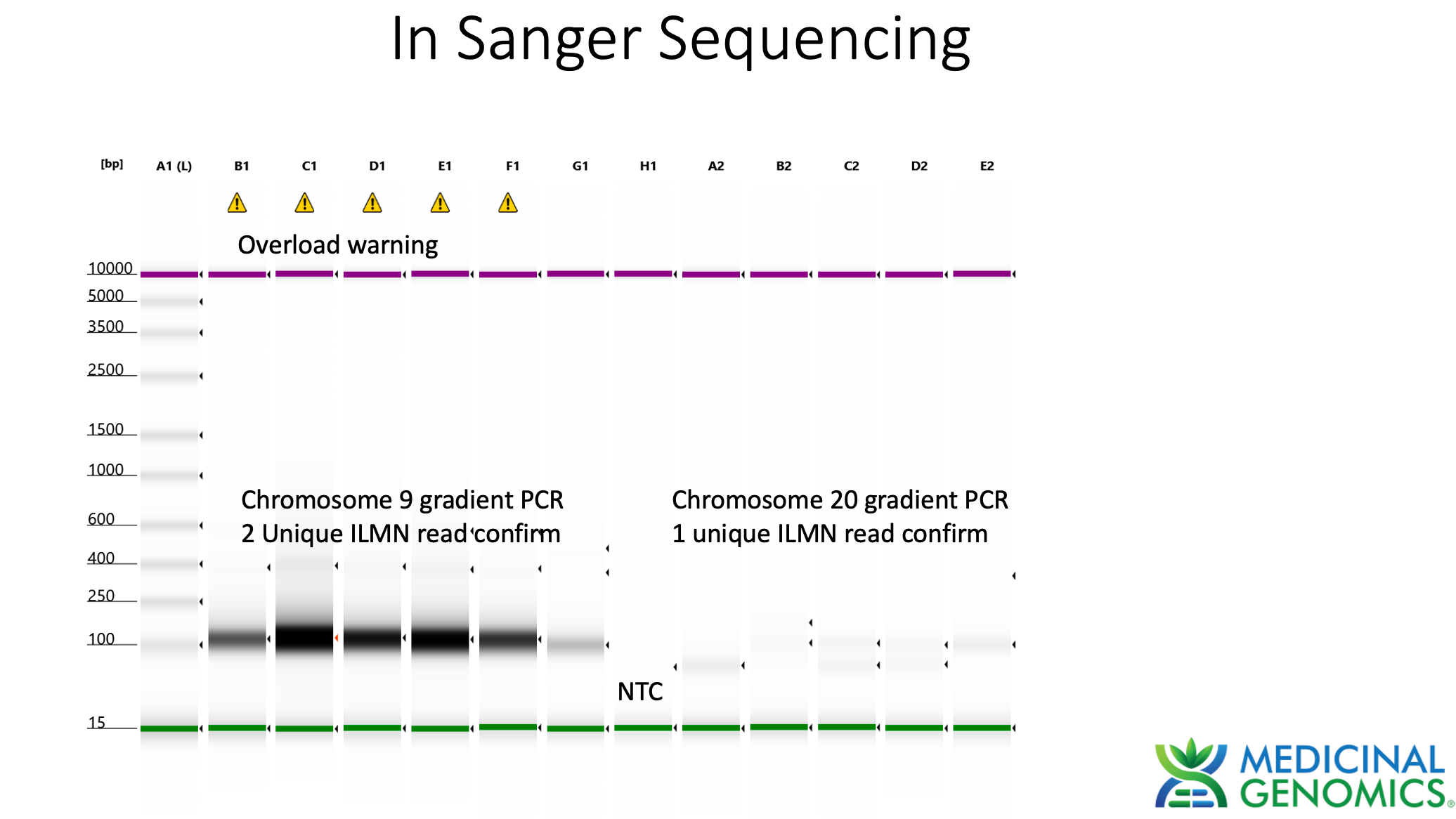
Sanger verification of the integration event in the Illumina Library is seen above.








Additional observations
Plasmid DNA replication in Cell lines
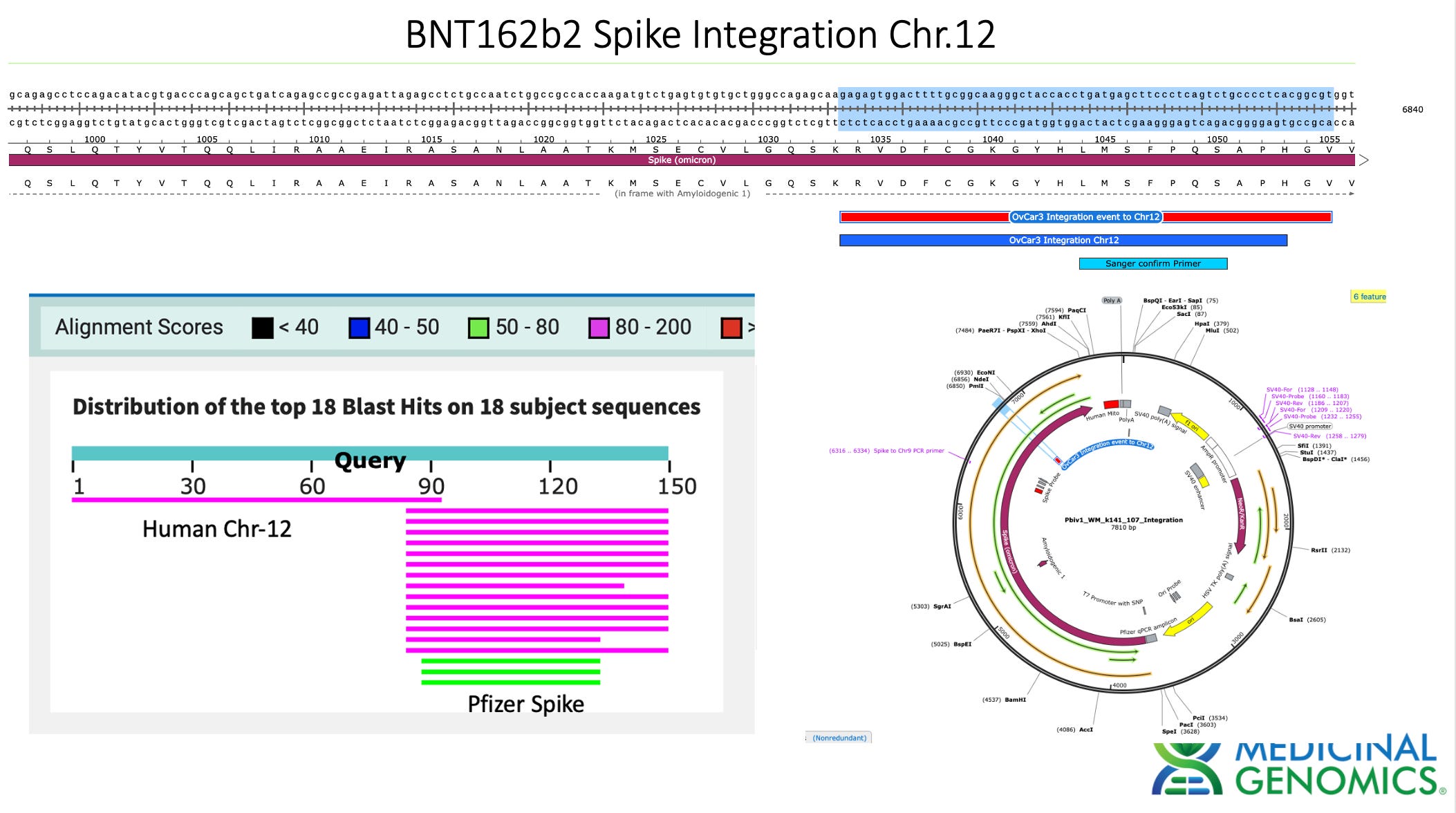
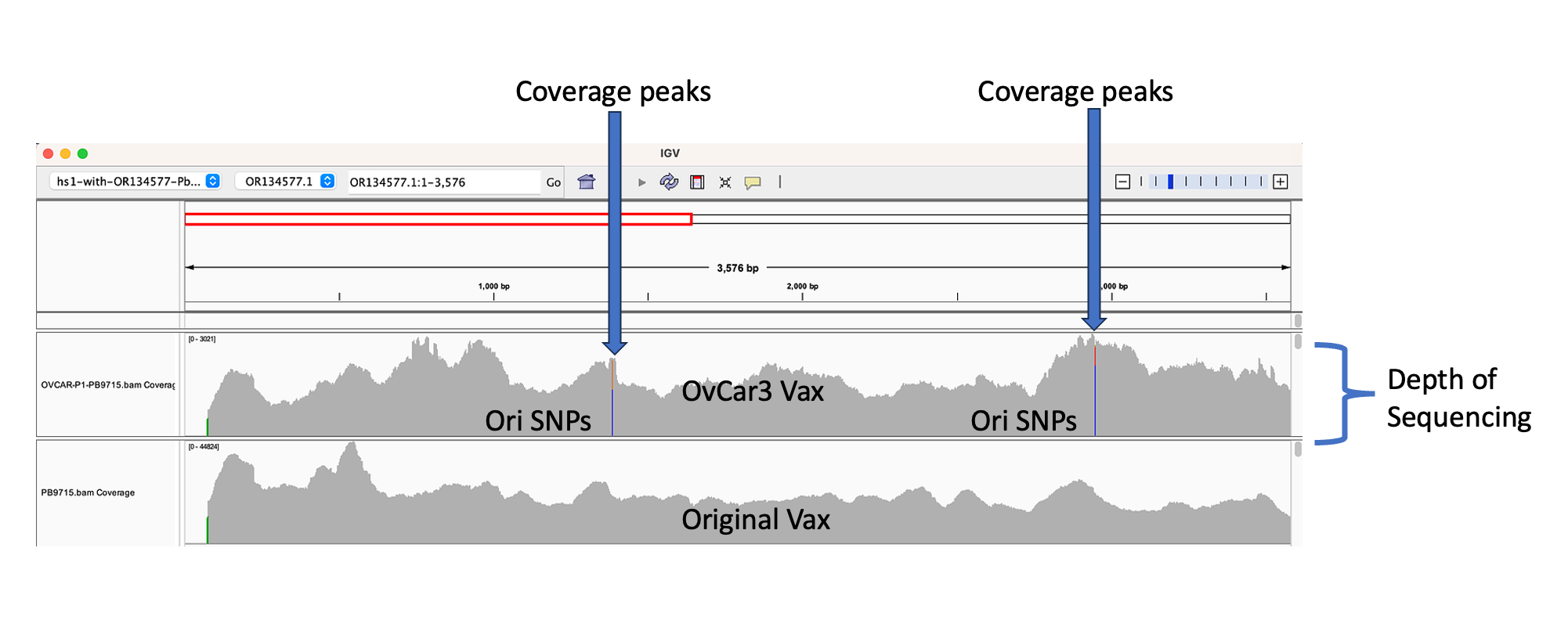
The depth of sequencing over the vaccine in OvCar3 is over 3,000X while the depth of sequencing over the OvCar3 genomes is 30X. As a comparison, when sequencing a diploid human genomes (2X coverage) you usually get 1,000-10,000X coverage for the mitochondrial genome as it is multi copy in each cell. A similar thing is seen in plants that have both mitochondria and chloroplast genomes. These vaccine plasmids are at 100X coverage per 1X human genome. That is higher than I expected and far higher than any fact checker predicted.
Another odd thing is observed. There are SNPs in the plasmid vector that do not appear in the vaccine that is never exposed to any cells. This is a sign that the cells are mutating the plasmids.
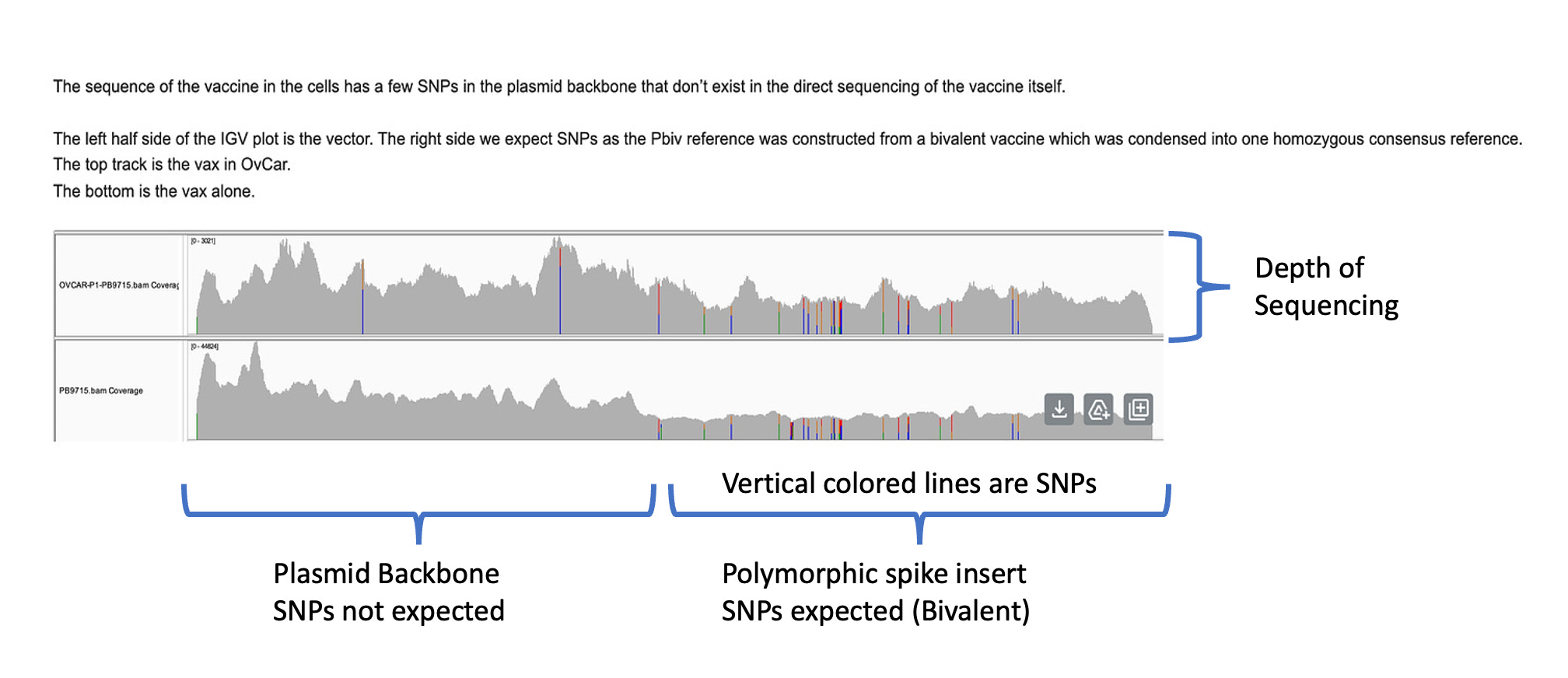
Why are there SNPs in the plasmid Origin of replication only when the plasmid is transfected into a cell? These SNPs are not present in the vaccine control sequence (Left side of bottom track in above figure). The variants are in phase and are 12%-40% in allele frequency.
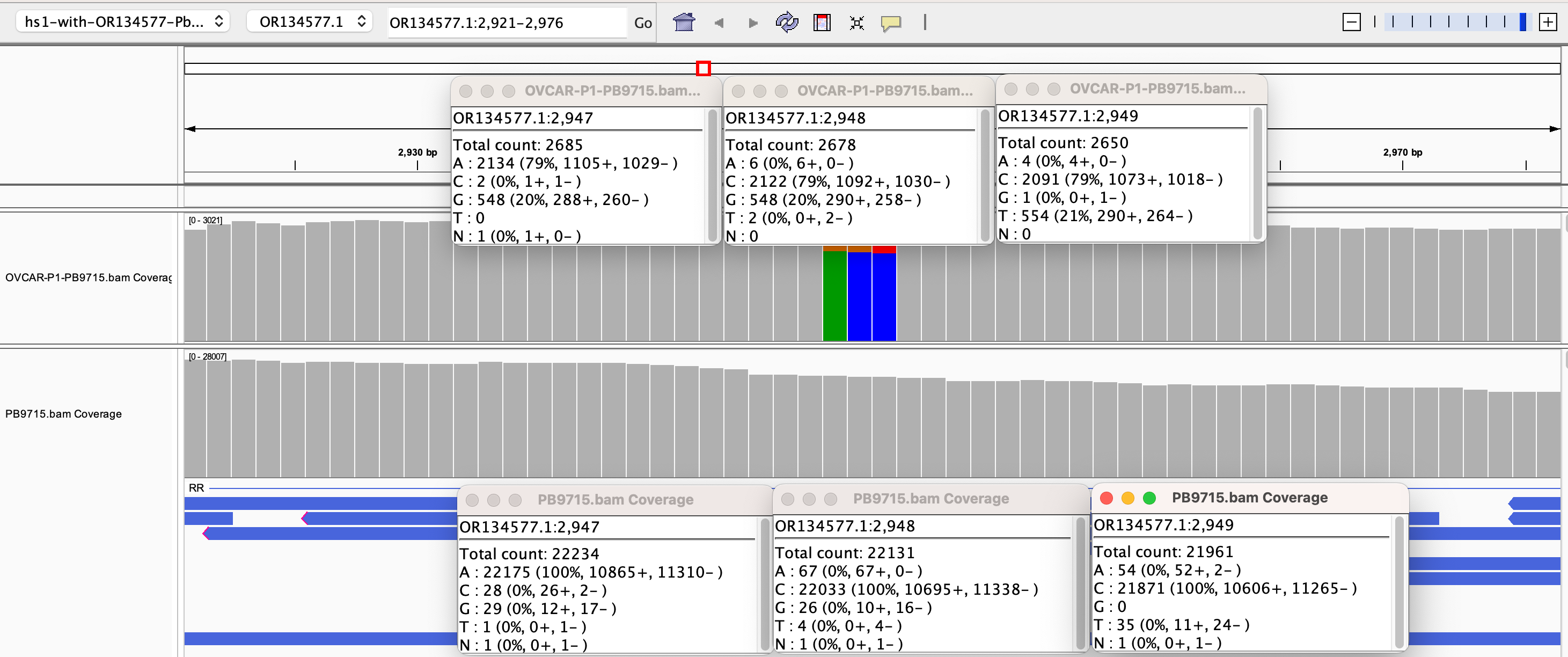
If you lower the allele frequency to 10%, one more SNP appears in this Bacterial origin of replication.

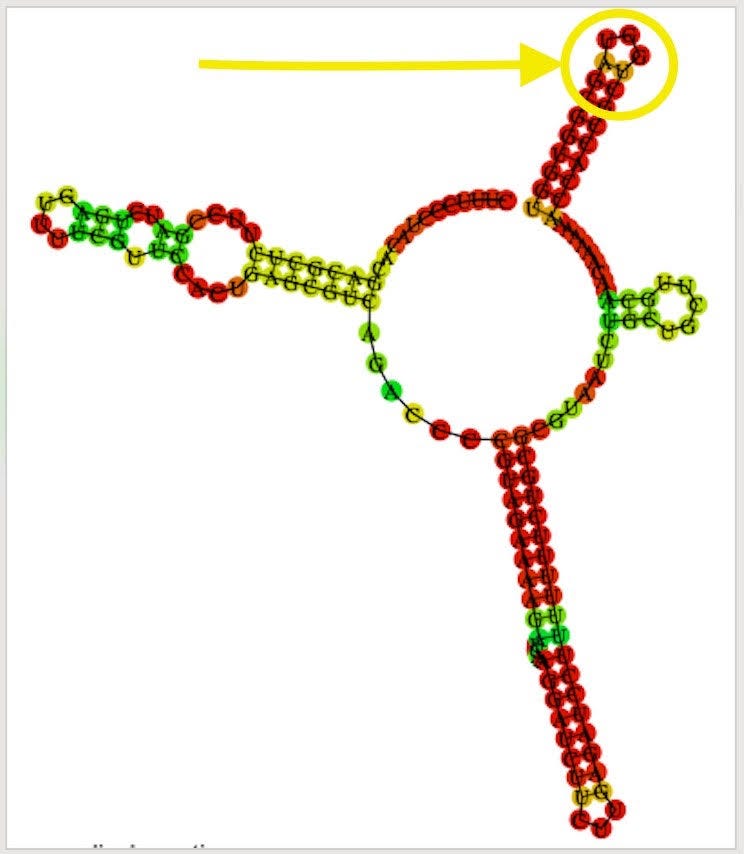
There is also a SNP in the F1 Origin of replication (12% allele frequency)
The F1 Origin is derived from a Phagemid which initiates ssDNA synthesis. They are known to form hairpins.
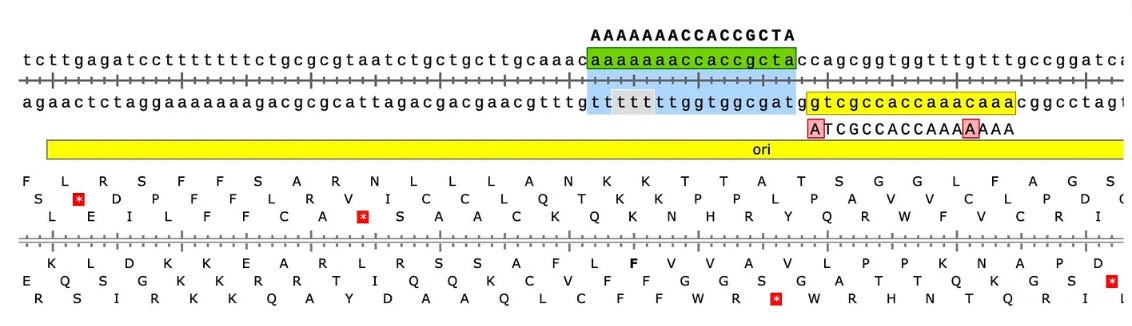
The variants are in the stem loop of the Bacterial Ori highlighted in yellow.
There is a fifth SNP in the OvCar3 vaccine sequence at the 3’ end of this highlighted stretch in the SV40 Origin (GAGGC[C/G]). This is at 40% allele frequency.
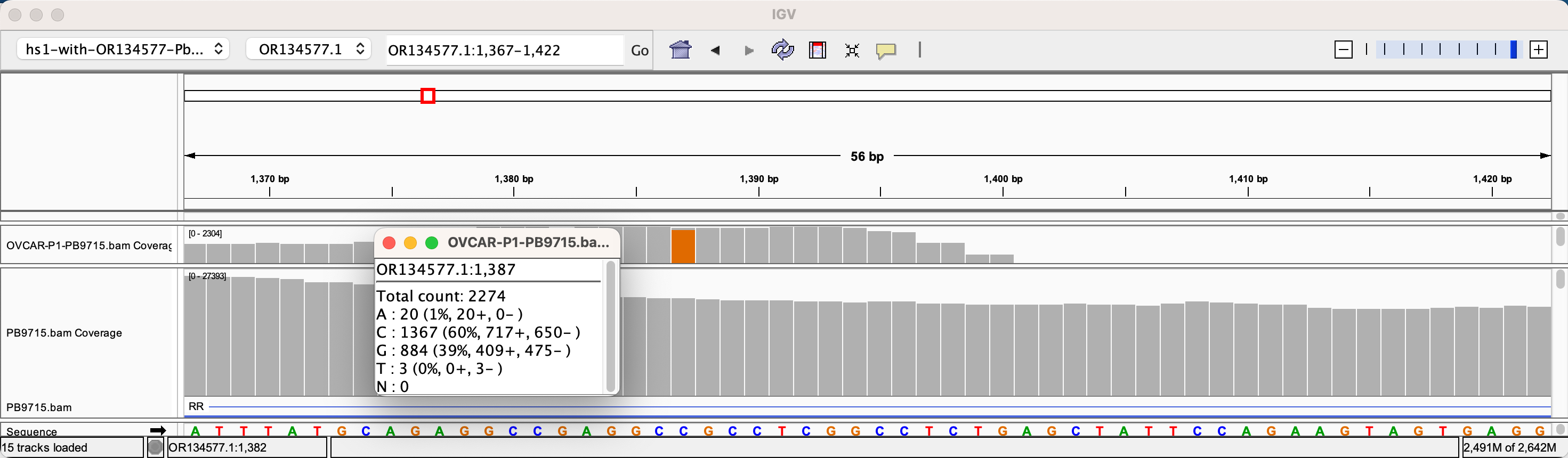
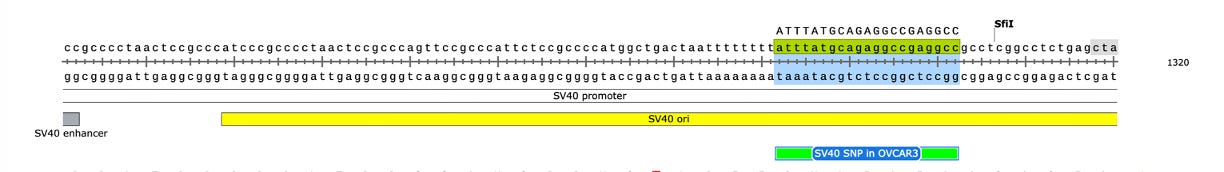
Given all 5 of these variants exist in Origins of replication, this is likely evidence that these components are being replicated in OvCar3 cells lines and these are replication errors. This is supported by the higher than average sequence coverage over these regions of the OvCar3/vaccine sequence. Note the SNPs fall on coverage peaks in the IGV coverage plots below. The replication is unlikely to span large distances as most of the fragments are short in length but some of the larger fragments observed with oxford nanopore sequencing of the vaccine could be readily amplified to higher levels once inside the cell. Some of these 3kb fragments contained the entire Neo/Kan gene with its SV40 promoter and SV40 Ori.
Discussions and Limitations
This is a cancer cell line that doubles every 60 hours. This doesn’t imply that this is happening in patient cells but it may be a model for what a cancer patient in remission may face upon vaccination.
There are several limitations to this study.
1)While we have Sanger sequence confirmation of these chimeric events in the Illumina sequencing library that was generated, we consumed most of the cells to make that sequencing library and cannot confirm this is in the remnant cells we have. Future studies will culture larger quantities of cells to afford more retrospective scrutiny of these events in the original vaccinated cell line. These are rare events so they are unlikely to be present in all cells and resampling a different collection of cells from the same batch of cells may not capture the same events.
2)Since we used short reads from ILMN, we do not have confirmation of both ends of the integration event. Thus we cannot opine on the size of the integration event until we capture the other end of the integration event. Deeper sequencing of the Illumina library may find this event and the use of longer reads (ONT or PacBio) should be explored in future studies. These platforms require more DNA input and may require a scaled up transfection (more cells).
3)Both (Chr9 and Chr12) putative integration events are supported by multiple reads in the library. Singleton events cannot be trusted as they could be the result of chimeric ligation in the Illumina library ligation step. The odds of two molecules in the library sharing the same integration point as a function of random chimera formation in library construction is 1 in 3 billion likelihood. Having this occur twice (Chr9 and Chr12) in the vaccinated cell lines with 2 independent reads supporting each event and zero times in the unvaccinated control implies these are strong contenders for integration. Sanger sequencing confirmation of amplicons traversing the integration event using different primers than used on the Illumina cluster station, implies these are not artifacts of cluster PCR, index hopping or optical artifacts known to occur at low frequencies on the Illumina sequencers.
4)Cancer cell lines are known to go through Chromothripsis which is a shattering of the genome. It is possible these chimeric events represent ligation to fragments of chromosomes which are extrachromosomal. Therefore long reads are required to resolve more ephemeral chromothripsis events from longer term chromosomal integration.
In summary, this data does demonstrate that SV40, Origin of replication and spike DNA can be found through at least 2 cell passages in vaccinated OvCar3 cell lines. This is not seen in the untreated cell lines. Additionally spike DNA was detected in Whole Genome Shotgun libraries of the vaccine treated samples at 3,000X coverage while the human genome was only at 30X coverage. Further work is required to validate the size and frequency of these integration events and rule out Illumina related artifacts that could lead to false positive integration events.
Additionally, detection of SNPs in the vaccine plasmids origin of replications (F1 and SV40) is a sign that OvCar3 cells lines are initiating replication of DNA once transfected. Thus small amounts of DNA contamination containing superfluous mammalian origins of replication can result in larger quantities of DNA once inside a cell. This should inform regulations regarding residual DNA contamination. A simple 10ng limit on all DNA should be reconsidered if the contaminating DNA contains billions of copies of mammalian origins of replication. The Moderna vaccines do not contain SV40 origin or F1 origin thus it is obvious these sequences are unnecessary risks in the Pfizer vaccine.
This work has taught us that several regions of the plasmid DNA that are identical to human mitochondrial sequences or polyA sequences cannot be ascertained for integration as they exist in both the vaccine and the human genome.
Further work is required to validate these putative integration events. Longer reads and deeper sequencing are now justified and should be prioritized.
FastQ Sequencing Data for OVCAR3-PB9715
These reads are untrimmed and identical to the reads provided from the sequence provider.
•https://mega.nz/file/9ZgyjYrQ#zZUf8X5FkW-a2_sGBN4G37TqQkLFITMQf77_-Uq0ZAo
•https://mega.nz/file/VRQwyAiT#LQGQCjNv3YNqCImrH8ENJGdek5qxphGXS6xTZucAMII
We have updated this data store to contain the trimmed data as well. These hashes will not match those from the sequence provider as we have trimmed the reads per suggestion by Dr. Hiroshi Arakawa.
These contain whole genome sequencing of BNT162b2 treated and untreated OvCar3 cells.
https://mega.nz/folder/hFwSUaSJ#p1I9OwzBwzBsnXEHpPmfUw
https://mega.nz/folder/RFxxgATI#AU9Yey8IH5OAeMA-YeHx-A
If your browser asks for an encryption key, try another browser. The encryption key is after the # in the URL but some browsers don’t parse this.
Hoping someone will post a summary for dummies.
Do you think stables be created by selecting with neo, after serial passaging? You’d only get a subset of insertion events, but they could be maintained?