Layman's description of RNA:DNA hybrids
An intentional game of "Hide the Ball"

Our most recent paper on RNA:DNA hybrids stems from our Veterans Day Vials work.
This paper has been accepted with minor edits for publication. Ironically BioRXIV (a preprint server) censored the work.

The key take away from this work, is that the Regulators are “Digging in the wrong place” for the DNA and this is a very suspect plan as BioNtech has led them there deliberately.
1)BioNtech has a Spike qPCR Assay described in their EMA documents. They never report the CT value this qPCR assay produces. They claim to only use it to prove the plasmid isn’t empty and in fact has a spike insert.
Proof- Go to Page 80-81 of this EMA document.
You will find this spike qPCR assay.

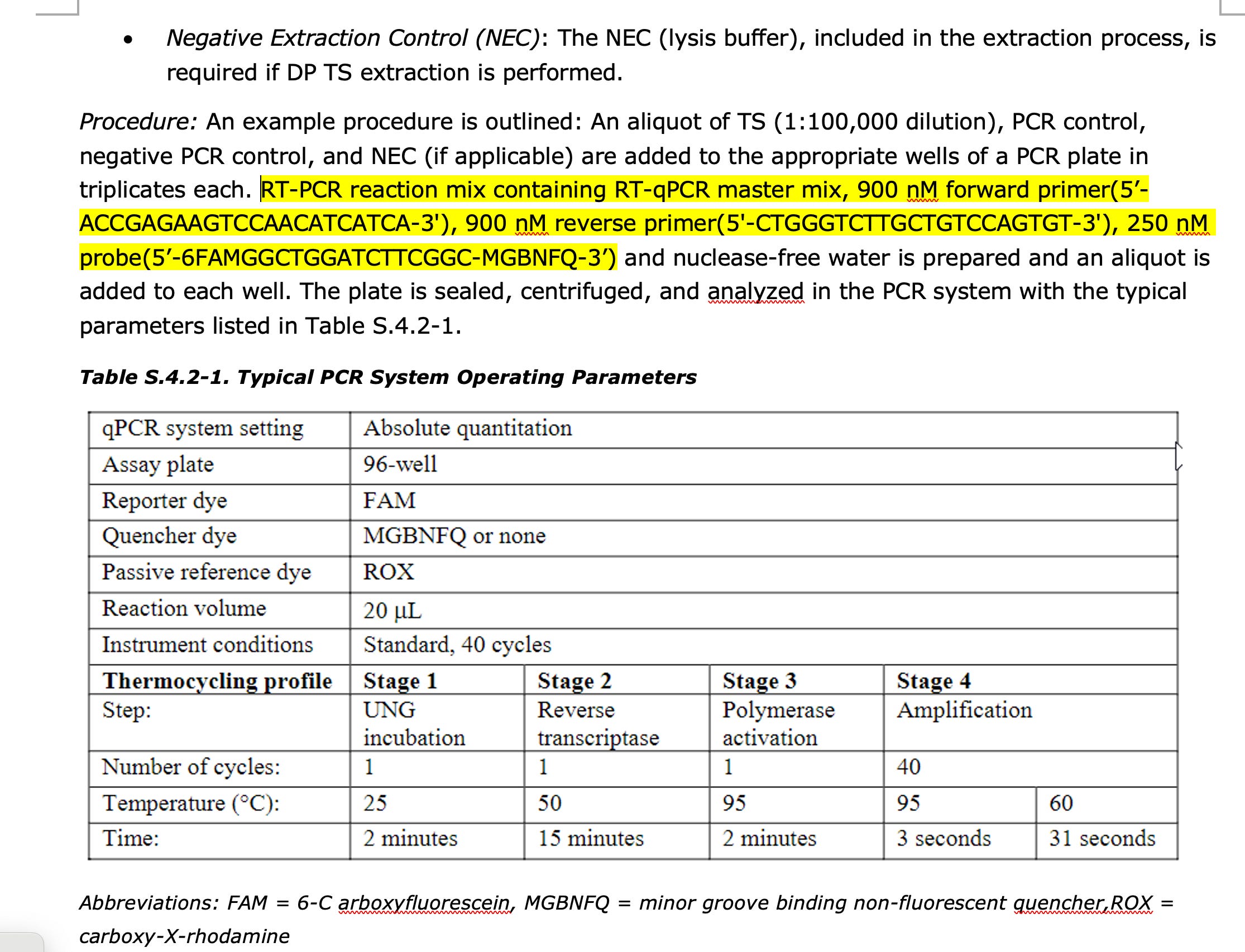
If you load those sequences into SNAPgene with the Pfizer reference they will confirm that they are targeting the Spike gene.
There acceptance criteria is only if this assay is below CT 32 and their NTCs are greater than CT 32. But they will not share this CT value because if they did it would reveal their 100X over the DNA limit issue.
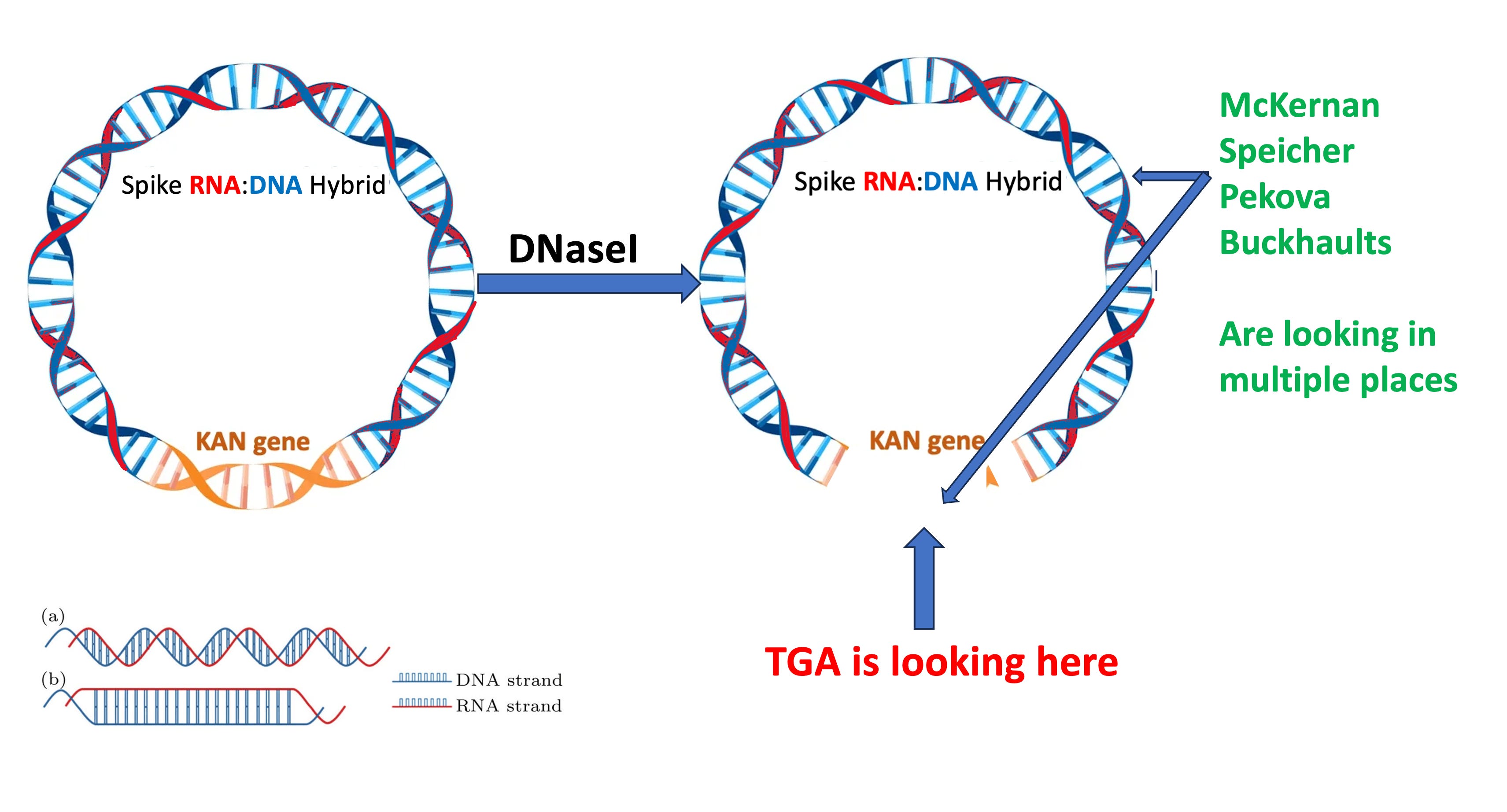
2)BioNtech is fully aware that the DNaseI they are using will not erase the spike DNA. This is from their Lenk et al paper. Yes.. This is a paper from their scientists so they cannot claim to not know this well known property of DNaseI. Sutton et al documented this in 1997.

So BioNtech has a Spike qPCR assay. They use it. They have it validated. They are aware that RNA:DNA hybrids are not digested by DNaseI. So instead of using the CT value from an already validated test, they invent a new qPCR test that looks the other way.
Where do they point the regulators? This is from the heavily redacted TGA document. The TGA claims everything is fine because they have checked the KAN gene, not the Spike gene.

The KAN gene is not protected by the modRNA after the IVT reaction.
That brings us to the 1st Figure on our Paper and how this will impact qPCR estimation of the residual plasmid DNA. If you only look at a single 100bp region and attempt to extrapolate that reading to the rest of the 7824bp plasmid you are making assumptions that the rest of the plasmid exists in the exact same copy number as your small KAN target. Fatal assumption when you know DNaseI will not process the Spike part of the plasmid.
Another pointed question for the regulators? How did you get convinced to use the KAN assay? The clinical trial used Spike PCR products as IVT templates which had no KAN sequence? You literally got hoodwinked here. Why on earth would you use a qPCR assay that targets a region of the plasmid that is NOT amplified to measure the amplified DNA? Why would you do this when they had a Spike qPCR assay on record already?
Now when they switch to Process 2, the KAN assay might make sense if they were using the correct DNase to get rid ALL DNA. But again they are using the WRONG Dnase for this.
This is why there is so much disagreement on how much DNA is in these shots. Its NOT a Qubit cross talk problem when 1000X the amount of RNaseA is being used to get rid of the RNA. Check the units on our math. 20ug of RNaseA is a boat load to digest 1/300th of a 30ug Dose.
What does Buckhaults lab see.
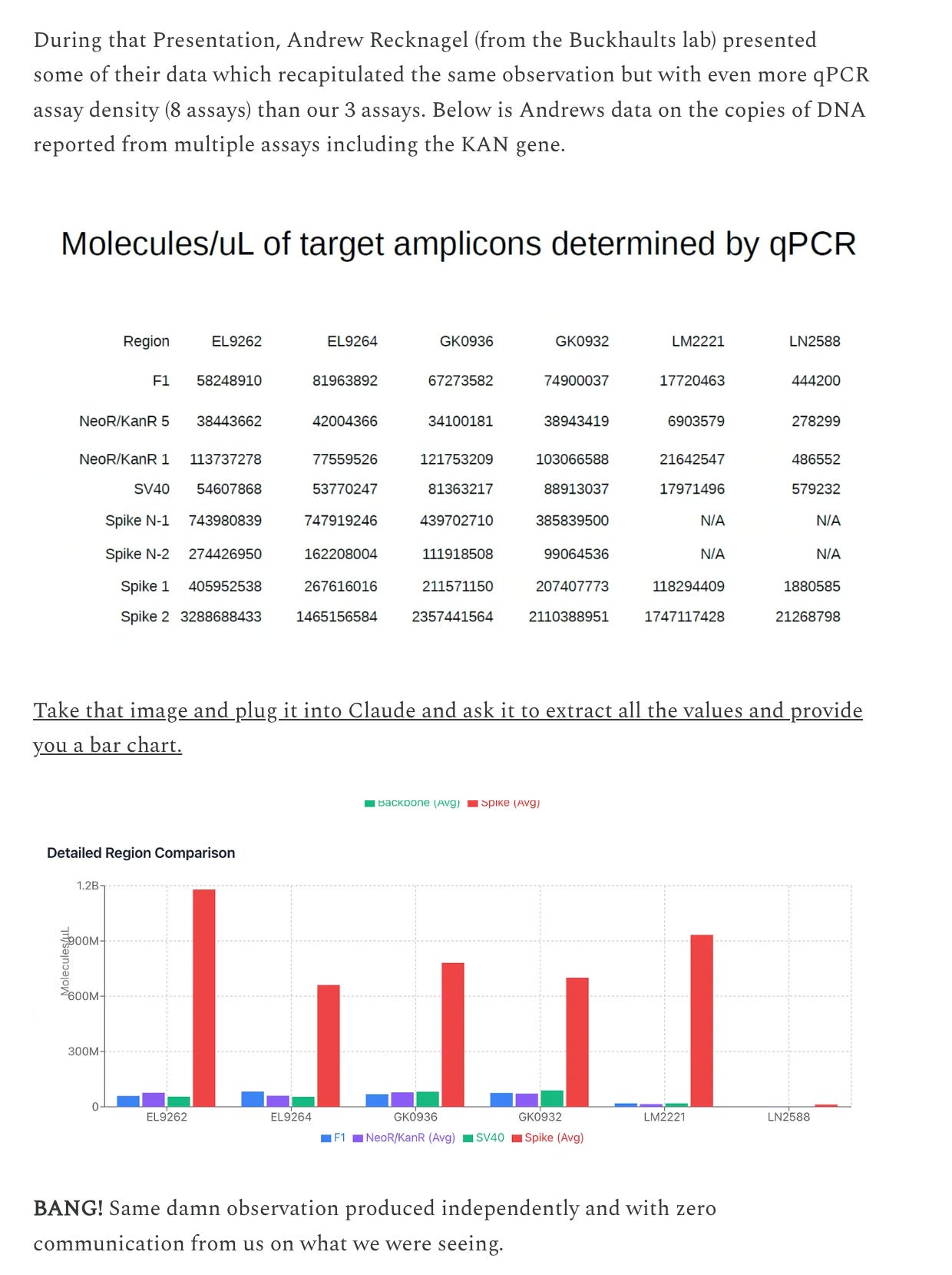
What does Sonia Pekova see?
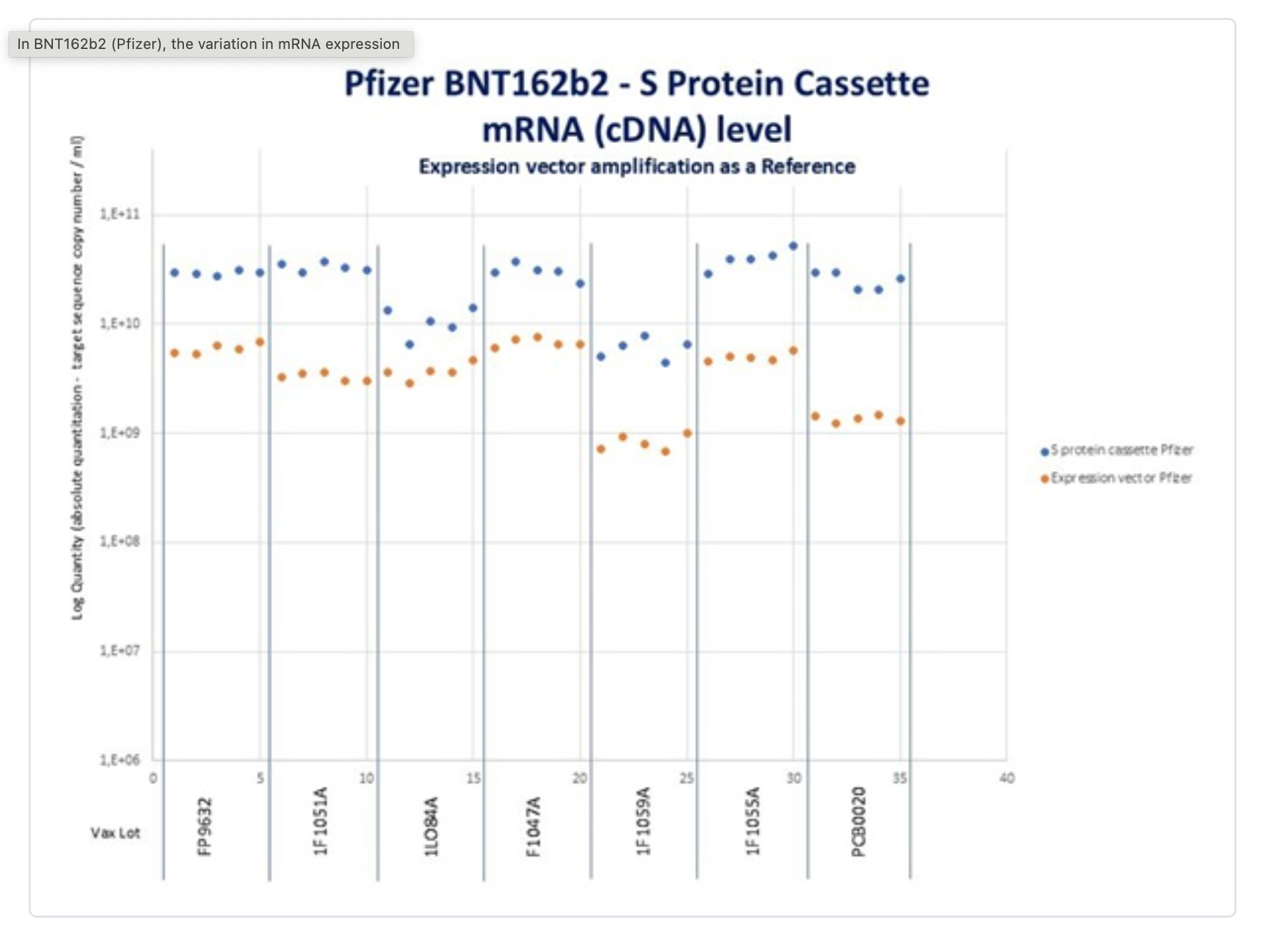
So two other labs are seeing the same problem. Orders of magnitude more Spike DNA than plasmid backbone.
How do prove the mechanism of action?
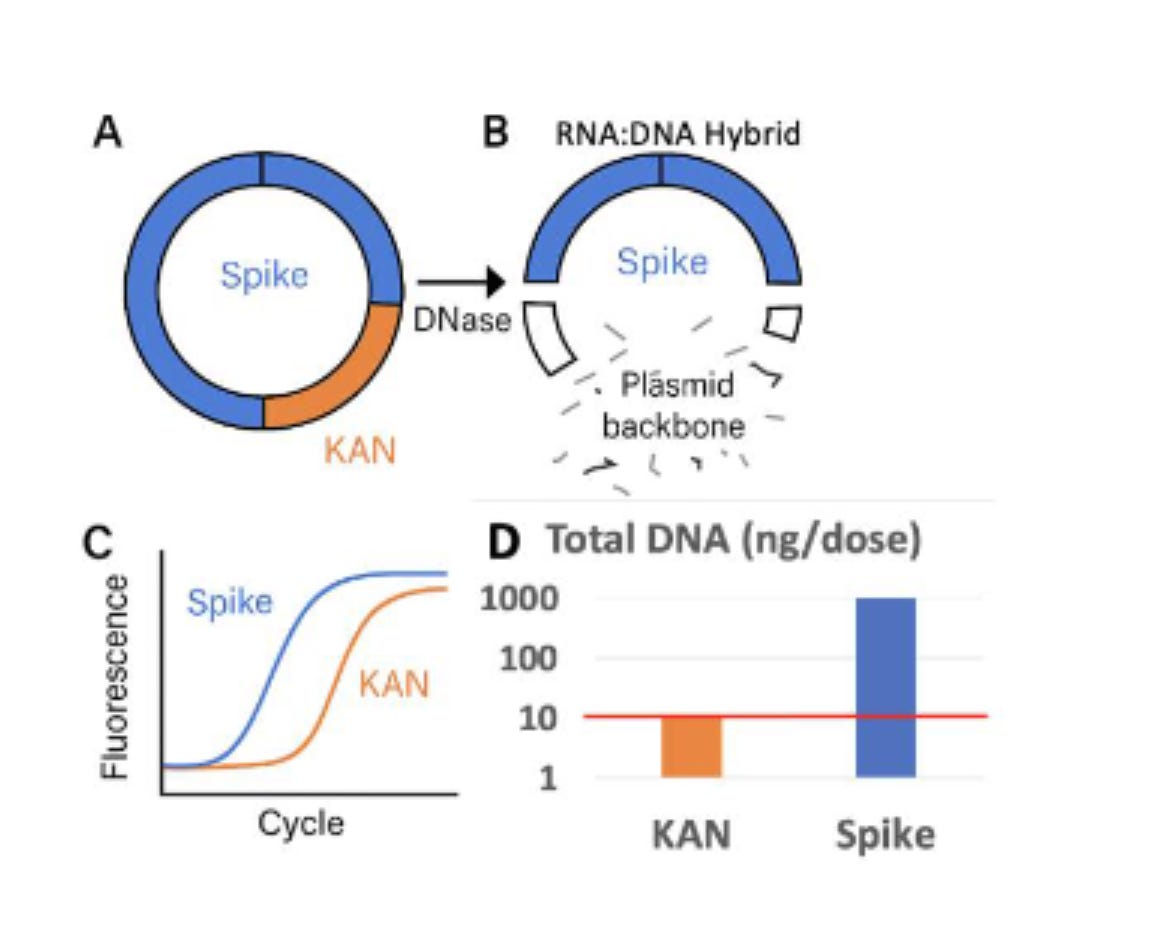
1)Take the existing Vaccines and break open the LNPs (TritonX-100) so they are Nuclease available.
2)Treat them again with DNaseI and qPCR the Spike vs Plasmid backbone (Ori)
3)Compare these to the same samples treated with an Enzyme designed to remove RNA:DNA hybrids (DNaseI-XT).
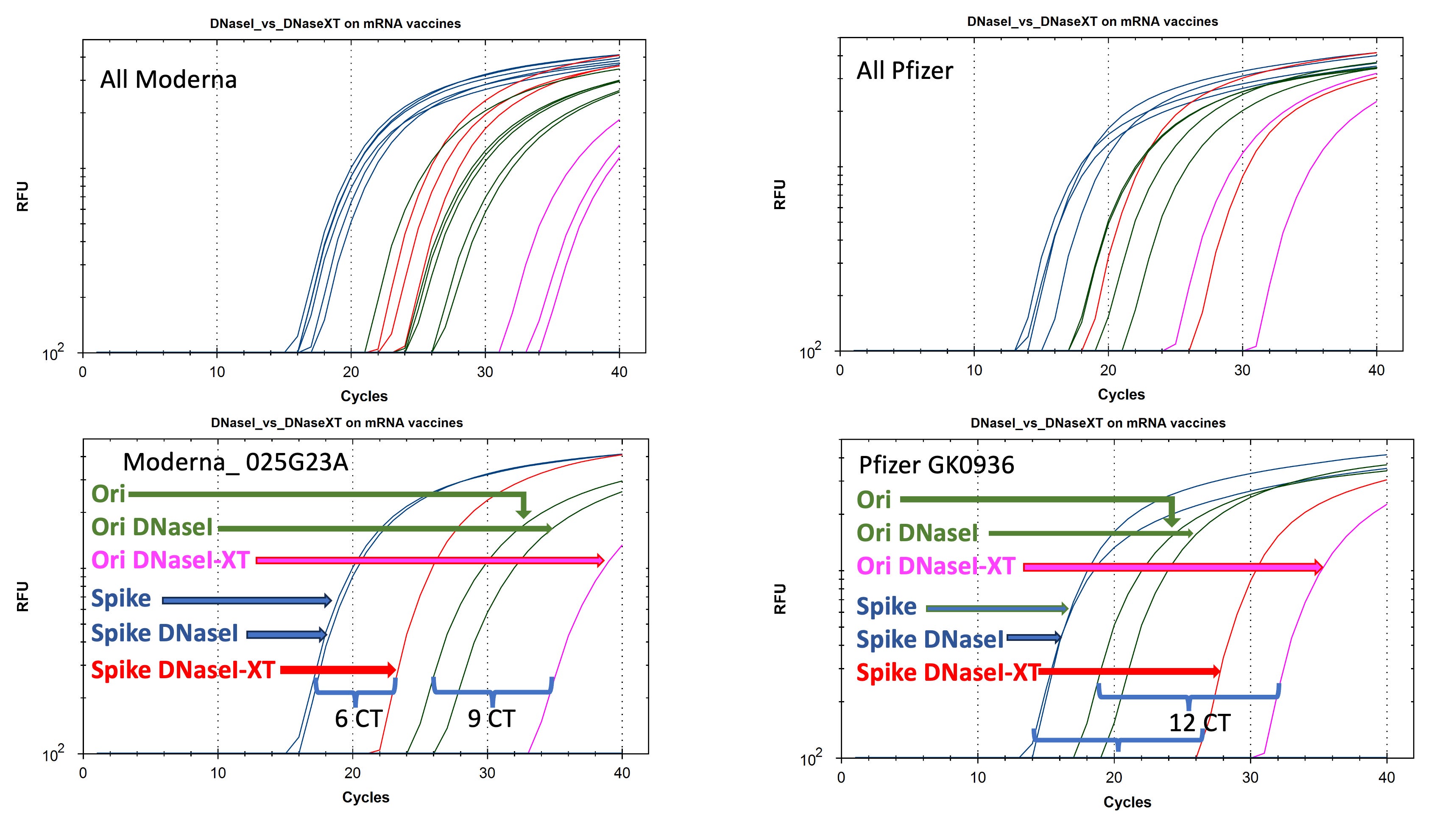
When DNaseI is applied to the Moderna vaccine the Spike signal in Blue doesn’t change. Only the Ori signal decays to a later CT.
When DNaseI-XT is used, Spike shifts 6CTs (100 fold) and Ori shifts 9CTs (1000 fold).
This why you need to use RNaseA Fluorometry which measures all DNA in the vial. Not some 100bp region that you model and assume to represent everything that is there.
Important video that allows you to see this live.

When you do this correctly the vials are all orders of magnitude over the limit.
A few caveats… Even RNaseA-Fluorometry will under measure DNase treated DNA by 70%. Georgiou et al documents this beautifully. This is due to shorter DNA not staining as well with the dyes than the same mass of DNA of longer length.

Now that we know that RNA:DNA hybrids are present this does invoke another question: how much ssDNA is present as our assays do not stain ssDNA?
For every RNA:DNA hybrid, there is one Crick strand (Sense strand that codes for spike) hybridized to one RNA strand.

The other Watson DNA strand is displaced and not measured with RNase A free Fluorometry with PicoGreen. It should be measured in qPCR as primers amplify both strands. After RNaseA treatment, the DNA strands should reanneal if the sample is heated to 95C and cooled to room temp in 10 minutes but this CoT curve may leave some ssDNA that goes undetected with Fluorometry. OliGreen dye would be worth testing to better measure both ssDNA and dsDNA contamination as both are stimulators of cGAS-STING.
Conclusions: This is well engineered Parlor Trick. Its a game of hide the ball. They are aware of this deception as they published about RNA:DNA hybrids failing to digest with the very DNaseI nuclease they use in manufacturing. They pointed the regulators at a region of the plasmid known to be removed while knowingly leaving spike intact. I say knowingly as they have a qPCR assay for Spike. They have run it and they know the CT value but they refuse to share it. Other independent scientists that measure these two regions see 6-7.34CT offsets which explains the 100 fold difference from independent researches vs Regulators.
Most Regulators are not running this qPCR test. They are trusting Pharma to produce honest numbers not realizing this “Validated Assay” is a complete scam. When challenged with independent data they resort to accusations that the independent assays are not “Validated”.
Never forget that as they criticize Fluorometry for having too much cross talk! They then resort to using Fluorometry to measure the dose of the RNA being injected. They use RiboGreen for this which has 10X higher cross talk to DNA than PicoGreen has to RNA (Jones et al). So this is a complete smoke grenade they propagated.

Why should you care about 100X more Spike DNA?
Well, that is the exact nucleic acid that is being detected in all of the RNA/DNA persistence data (Krauson et al, Castruita et al, Roltgen et al, Gonzalez et al, Hanna et al, Ota et al). Most of these studies are using RT-qPCR which amplifies both DNA and RNA and no study to date has used a proper nuclease to different these two forms of nucleic acid and reporting persistence data as if it is RNA.
And this spike DNA has a Cryptic Mammalian promoter Pfizer snuck into the design. That means it can express RNA in mammalian cells. This of course needs to be tested.
It’s not hard but our lab doesn’t have mammalian cell culture. If someone else does, this is a simple experiment to check.
1)Take the vaccine. RNaseA treat it. Repackage the Remaining DNA into Lipofectamine and transfect HEK293 cells.
2)crack open the cells,
3)qPCR SV40 or spike to confirm the DNA made it into the cells.
4)Treat with DNaseI-XT to eliminate the vax DNA.
5)RT-qPCR for SV40 and Spike.
This simple experiment will determine if this cryptic promoter is active in HEK293 cells.
If the residual DNA is making more spike RNA, that would explain persistent spike expression 700+ days out.
Cryptic Promoters

This has always been a question. Now that we have shown there is failed linearization in the Pfizer plasmid the next excuse we should expect delivered from the defenders of this manufacturing slop, is that T7 Promoters do no transcribe in Mammalian cells so even if plasmids stick around, they will be ‘inert’.