

Targeted Enrichment and sequencing of BNT162b2 from OvCar3 cells
Optimizing sequencer efficiency


Sequencing can be expensive if you are looking for needles in a haystack. Often diploid human genomes are simply sequenced 30-90X coverage to get adequate sequence coverage of both maternal and paternal chromosomes. This usually equates to 600M 150bp Illumina reads (90Gbases) for a 30X coverage 3Gb genome or a 2B reads for a 90X 3Gb genome.
To sequence a diploid human genome, collections of 100,000s of cells are bulk DNA purified and sequenced with the assumption of the genomes from each cell being identical. This is a key assumption that doesn’t always hold true in cancer tissues, mosaic individuals or when you are hunting for variants that exist in only a subset of the 100,000 cells.
In the world of hunting for DNA integration, a small percentage of the cells are expected to be integrated and each integration event is likely to be unique to each cell. In these circumstances much higher sequencing coverage (10-100X more than a simple 90X coverage) is required to find such rare events (900-9,000 X coverrage). One publication made rather naive assumptions declaring no C19 viral integration exists after sequencing only to 54X coverage in diploid cells. This paper is less credible than the work performed by Rudi Jaenisch’s lab at the Whitehead Institute/MIT (Zhang et al) that employed methods to find rare events.
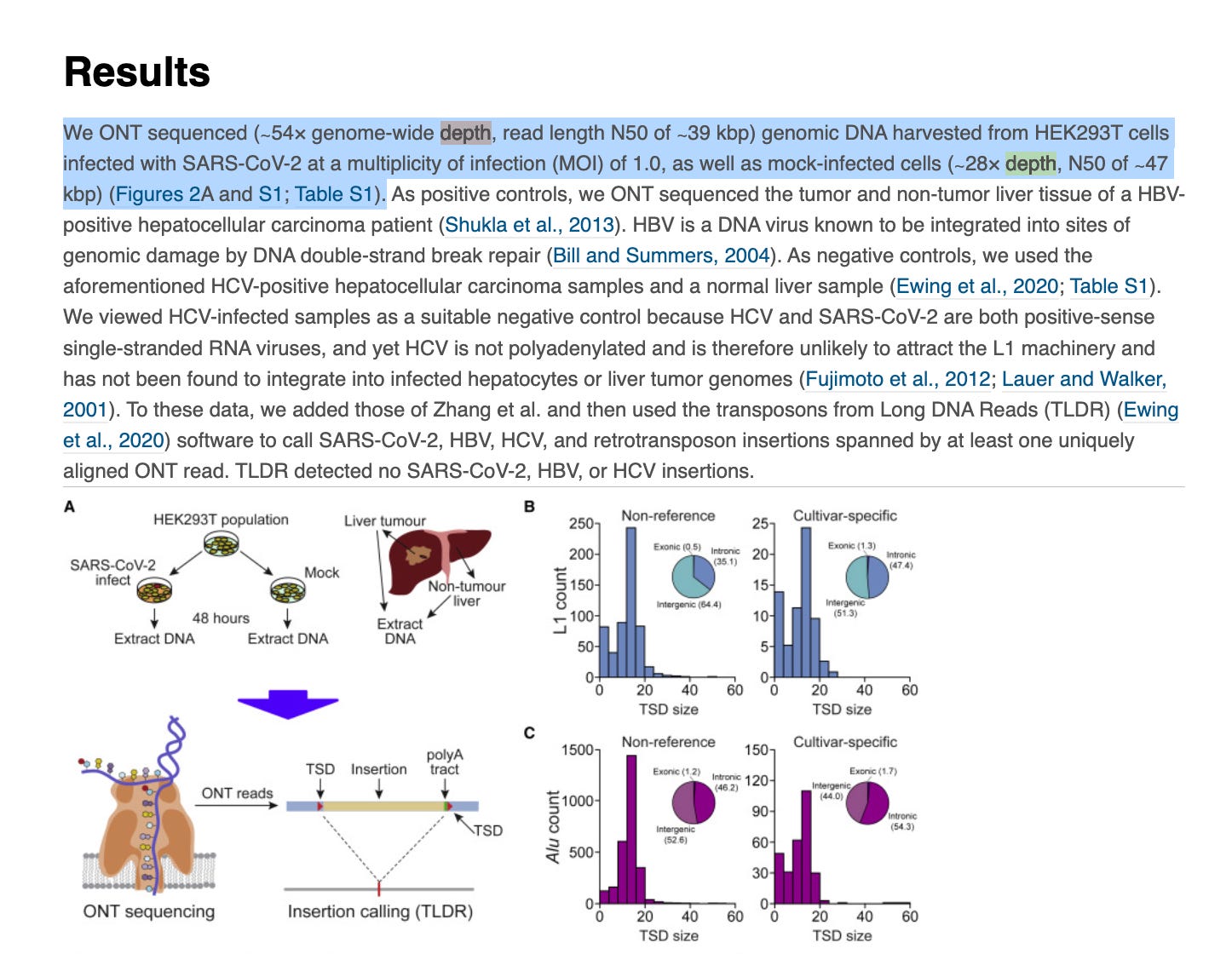
Zhang et al used a method called TagMap (Stern et al) to find rare SARs-CoV-2 integration events into human cells. This looks like a very sensitive method but appears to require custom assembly of transposomes and some optimization of the transposon integration frequency.
The method relies on PCR, however, PCR requires 2 primers to amplify. When you have an integration event that could occur anywhere in the genome, what primers do you use? One primer has to be human and one primer has to be from the vaccine.
Ideally, you would use multiple primers from the vaccine as you can’t be certain which piece of the vaccine integrated nor where in the human genome it integrated.
TagMap, tags the genome with a transposon bombing reaction. If you integrate a transposon every ~2000 bases in the genome, you can use the transposon sequence as a PCR primer site and a spike primer as your 2nd primer in PCR.
You can’t over transposon bomb the genome (ie. 1 event ever 200bp) as you will just amplify transposon to transposon, so much care has to be taken to keep the transposon bombing frequency larger than the distance your PCR is optimized to amplify in hopes of only getting transposon to integration sites that can amplify.
Its a clever technique but use of a single spike primer will only find integration events from that region of spike and as a result it will have many blind spots.
Now that the entire vaccine plasmid sequence is known, we designed an alternative approach described in a previous article. This approach may also be used to enhance the TagMap approach.

Briefly this method tiles the plasmid sequence with 113 biotinylated ‘bait’ probes that can hybridize any molecule out of the patient sample that has sequence similarity to the plasmid. These bait primers have to not hybridize to themselves so they cannot capture inverted repeats in the plasmid like the SV40 72bp enhancer.
To test this method we made a Whole Genome Shotgun library using a modified Watchmaker library construction kit. The one modification we employed was to add an additional 30 minute End Repair step (NEBnext End Repair kit #E6050S) to ensure the genomic DNA was fully end repaired and didn’t form chimeric ligations in the process of making Illumina Libraries. This approach is ultimately limited by the purity of the dATP used in the End Repair step. Any contamination with dTTP will result in T overhangs on the gDNA which can form chimeric fragments and chimeric reads.
Chimeric molecules in the library construction process were discussed in detail on previous threads. The % of chimeric reads is often noted on various library construction kits and will be an important topic as we try to assess the data that is enriched for BNT162b2 sequence.
For more information on library construction artifacts here are a few references. and Specs from Watchmaker
Results
The remainder of the BNT162b2 treated OvCar3 cells was converted into a whole genome shotgun library. This was sequenced to compare the number of reads that match BNT162b2 with and without the enrichment process.
We used the 16hr hybridization recommended for small targets in the IDT Lockdown protocol.
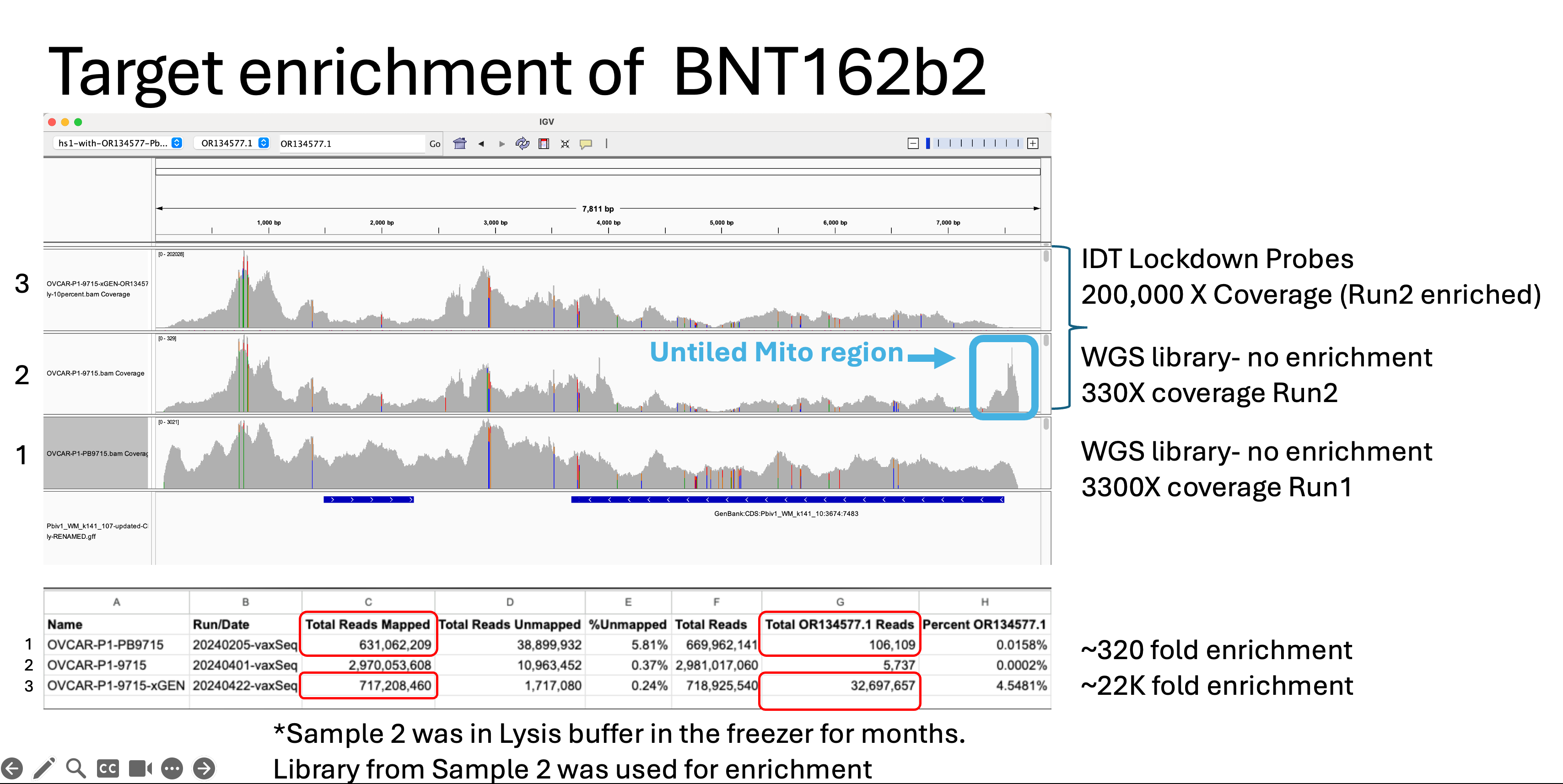
The coverage maps for the Run2 with enrichment (Top track sample 3) is nearly identical in shape as the unenriched sample (Middle track sample 2), how ever the coverage depth is ~600 time higher. Similar SNPs are seen in the plasmid (these are not seen when we sequence vaccines that have not touched cells). There is a small region of the plasmid that contained Mito Sequence on the right hand side of the middle track 2 (boxed in blue) that we didn’t tile with probes and hence doesn’t come through in the enrichment data.
We can see from the table above there are 2.9B reads in the unenriched sample and this only delivered 5.7K plasmid reads. Once the sample is enriched we have 717M reads with 32.7M plasmid reads. This is a 22,000 fold enrichment.
We did sequence this sample once before when the sample was freshly lysed and observed much higher plasmid coverage (106,000 X coverage). In the process of repeating this work to evaluate enrichment, we pulled those same lysed samples out of the freezer (after having been in lysis buffer for months) and made a new library out of the residuals from the tube. This delivered much lower (5.7K) plasmid coverage in the control but enriched to 32M reads.
Looking at this coverage in LOG scale provides demonstrates we are between 100,000X to 1M X coverage once the sample is enriched.
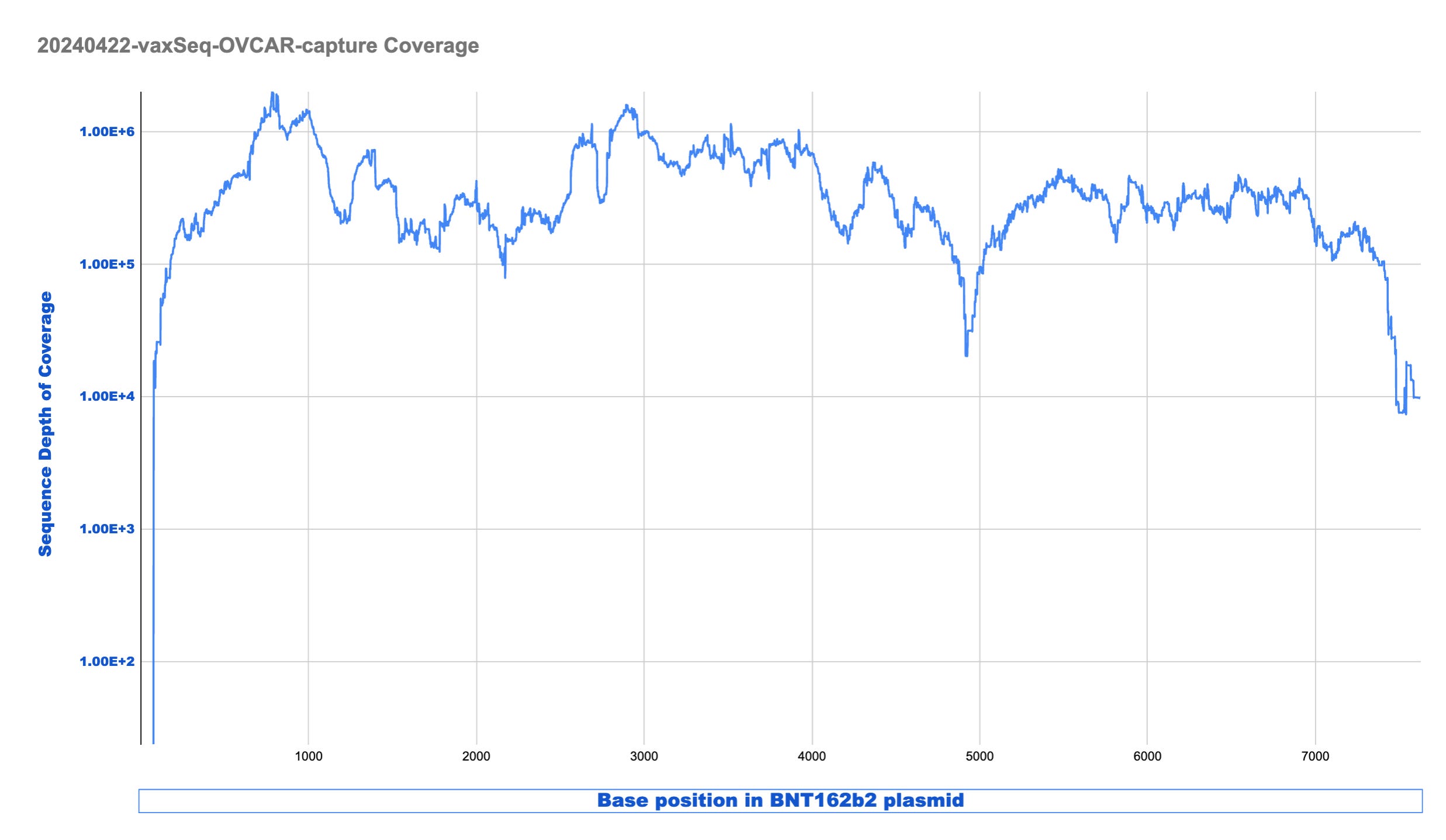
This is excellent enrichment and can reduce the sequencing costs required to find rare events significantly.
For those interested in these enriched reads you can find an excel sheet of all reads that map to BNT162b2 with one read and the OvCar3 genome with the other read here. Look a the 4th Tab over and you’ll see the data depicted in the table below.
You will note there are 18K read pairs in this 4th sheet and most of them are chimeric reads from the library construction process.
The reads we want to pay close attention to are reads that have a nearly full read length mapping to Human and a full read length mapping to BNT162b2.
You can identify the length of the mapping read by the CIGAR string in the column next to the blue box. 150M = 150bp Match. 4S146M = 4bp Softclip, 146bp Match. 146M4S = 146bp Match 4bp Softclip. These are alignment terms that depict how much of the read matches.
Of these reads which match near perfectly to each target we want to focus on loci that have reads mapping to the same general location with different start and stop points.
This means the 1st base and the last in the read which cover a loci of interest are offset compared to other reads covering the same region of interest.
This delivers you read pairs that are derived from different DNA molecules and can’t be chimera artifacts from the library construction process. Our previous substack on this topic goes over this chimeric read artifact and how to sift it apart with reads that have multiple start and stop points.

This ends up delivering DNA that is from spike on one read captured by lockdown probes 65-67. This happens to be one part of spike that contains a GP120-HIV motif.
CAGAAGCTACCTGACACCTGGCGATAGCAGCAGCGGATGGACAGCTGGTGCCGCCGCTTACTATGTGTGCTACCTGCAGCCTAGAACCTTCCTGCTGAAGTACAACGAGAACGGCACCATCACCGACGCCGTGGATTGTGCTCTGGATCC

and Human DNA that maps to chromosome 10.
TACGTGGGTCGCTCACCCCCGTGTATCCTGACATGAAGACACCTCCCGTTAGGGTCTAACATACACCTCATATAGGAGAGCTCTTGCTGGCATCTGGCAGGTGCCCCTCTCGGACAAAGCTTCCAGATGAAAGAACAGGCAGCACTCTTT

This maps to a region upstream of a lincRNA and down stream of an ENCODE Cis-regulatory element (CRE). See the center black block called YourSeq in the image below.

Part of this human sequence is in a repetitive region that is conserved in Rhesus monkeys. The lincRNA in the neighborhood is a ‘gene' or transcript known as ENSG00000288526 (right upper green arrows)

This is also known as transcript uc057vaa.1 and its linked to some thrombocytopenia phenotypes.
A more intuitive portal for understanding which regions of the genome are associated with which diseases is here.
https://hugeamp.org/region.html?chr=chr10&end=95168425&phenotype=T2D&start=95141925
Before we get too excited about these findings, it should be recognized that these are cancer cell lines given 100ul of Vaccine (1/3rd a dose) and we are not at a stage where we have eliminated all potential sequencing artifacts or confirmed these integration events with independent PCR in the original cells. We are in the process of ironing out all of the detection artifacts before we deploy this on real samples. Cell lines are the best place to sharpen that knife and we shouldn’t draw too many conclusions on integration rates with these data.
We have one more optimization that should occur. We have 18K chimeric BNT162b2 reads out of 717M) and a only a few that are candidates for integration events (have multiple start and stop point read pairs that support the event).
One way to clean up the chimeric read rate is to switch to a library construction process that is known to reduce these chimeric read events.
The current library construction process has a 3 step procedure where DNA is fragmented with DNase, End Repaired with a polymerase +dATP and then Adaptor ligated for sequencing. If this End Repair step fails for a few molecules, we can get genomic DNA ligating to plasmid DNA fragments thus creating false integration events. Since PCR is used after this step, these molecules can be amplified to create the false appearance of many reads (with the same start and stop points) that support a integration event.
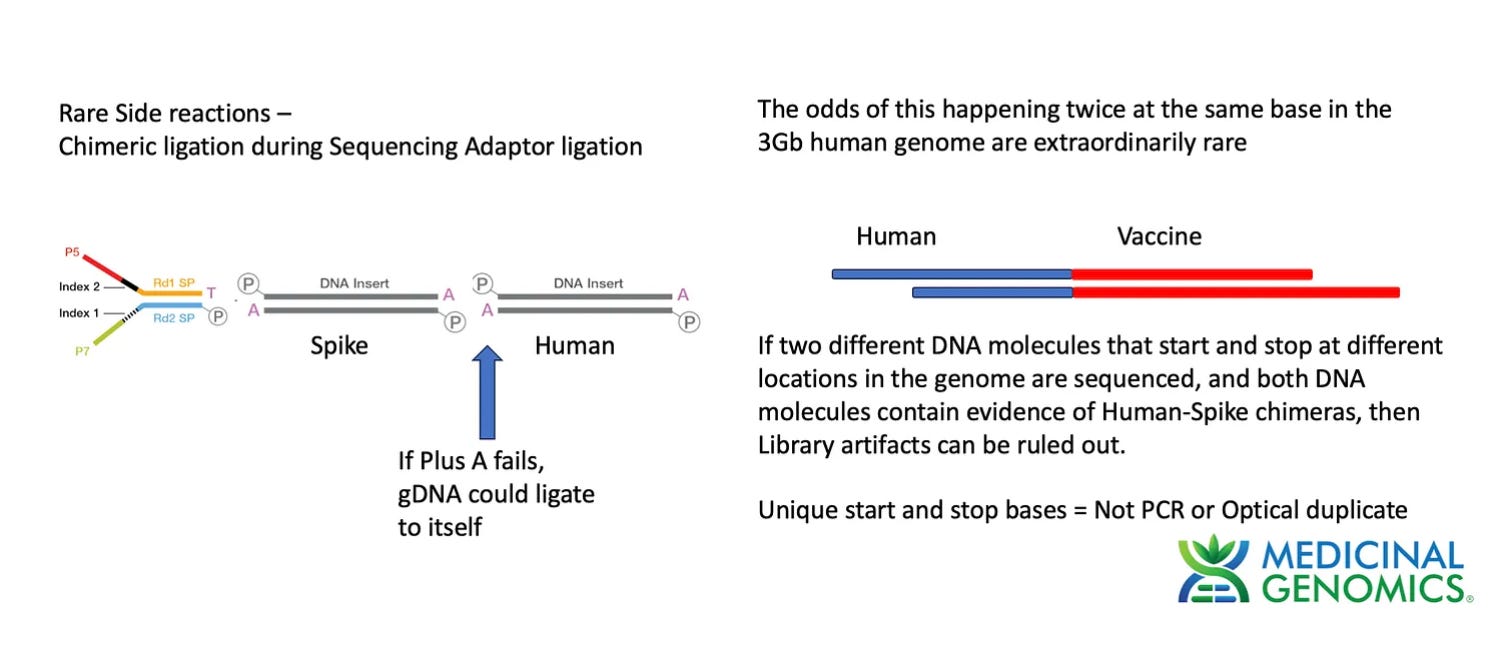
This is why we demand multiple unique or distinct molecules (unique start and stop sites) supporting an integration event. Reads that have unique start and stop bases inform us that they were derived from different molecules and the odds of chimeric read formation occurring at the neighboring base in the genome is 1 in 3 billion. Unique start points are one mechanism of eliminating the noise from chimeric reads but there is a cleaner way to do this.
The solution to reducing the amount of chimeric read formation is to use enzymes that perform the fragmentation and ligation steps in a single enzymatic step. Transposons are ideal for this.
A process known as Tagmentation can achieve this and is known to reduce chimeric read events 10-100 fold. This is what Zhang et al used with their TagMap approach.

Now that we have the enrichment giving us a 22,000 fold boost in targeting putative integration events, we are also enriching for any BNT162b2 chimeric reads (artifacts) in the Library construction methods. These are library construction artifacts we can reduce with other methods.
We can clean this up 10-100 fold by using a library construction approach less prone to chimeric read formation. Instead of 18,000 read pairs to shift through, the Nextera transposon based library construction process should bring this down to 180-1,800.

Target enrichment of Transposon derived libraries should be able to reduce the required reads to detect integration events from ~50 Billion reads to just 100M-500M reads. This is similar to Zhang et al use of transposons but instead of using a single PCR primer to search for spike integrations, we are searching for all possible vaccine derived integration events. Recall, Zhang et al published before the vaccine plasmid sequence was known.
This should enable dozens of patients to be screened on a single Illumina lane.
Stay tuned
Now that we have the enrichment techniques optimized we should be able to screen samples that are more likely to find integration events.
One must keep in mind that we are currently screening a vaccinated cancer cell-line culture as an optimization sandbox and there is a survivor bias. The cells that were harmed by integration likely die and we fail to sequence them. Since they are cancer cells before we even apply vaccines, we cant expect the integration events to grow much faster than cells that already double very quickly.
If we sequence actual patient tumors with such an approach, we will flip this survivor bias on its head as we will be looking in cells that were vaccinated AND then transformed into fast growing cancer cells post vaccination. This should be more fertile ground to find integration events compared to a cell passage of cells that were already cancerous pre-vaccination where the most mutated cells likely die and fail to sequence.
These are 30Gb Fastq and BAM files. If you are interested in lightweight data, the 9mb Excel sheet is the best place to start.
Data files
https://mega.nz/file/QERTGKgB#A7uDjBYZQoeRRFWZ0tiYgHssVsVQf4BGh9q9w3hwvnA
OVCAR-P1-9715-xGEN_R1_001.fastq.gz
https://mega.nz/file/kEZwRIga#ZViCp_EFKgMOTdoikXAvIGx4g3CJ0vp1t9iIvJ5QLQ4
OVCAR-P1-9715-xGEN_R2_001.fastq.gz
https://mega.nz/file/NB4G3LhT#qT-OnPUY60f6susead05X2tIIvExWI7Rk9MrznS_M3I
https://mega.nz/file/BYhFXZZD#p5pccZj3ctuy8WwT1PwzM5YdoCO0obToL5knoFAz0uA
Kevin, if you had been on the "vaccine" development team, we wouldn't be having to deal with this debacle. Also, I looked up "Relentless", and your picture popped up.
Thanks so much for all you do.
Don’t have a clue what you’re talking about but it sounds energetic and focused. Thank you Kevin. You really are a wonderful human being.